

 PDF
PDF Citation
Citation Print
Print



Introduction
Poly(ADP-ribose) polymerase inhibitors (PARPi) exhibit potent activity in germline BRCA1/2-mutated platinum-sensitive relapsed high-grade serous tubal/ovarian cancer (HGSC). In phase III clinical trials, maintenance therapy with PARPi was associated with a 73% reduction in the risk for disease progression or death as compared to placebo [1]. PARPi target the homologous recombination DNA repair defect (HRD) conferred by BRCA1/2 mutations, leading to tumor genomic instability and cell death. While germline BRCA1/2 mutations are detected in 15% HGSCs, genomic and functional data suggest the presence of HRD in approximately 50% HGSC [2]. The identification and validation of other HRD-associated biomarkers in sporadic tubal/ovarian cancer (OC) (hereafter referred to as OC) are crucial to potentially expand the number of women with OC who could benefit from DNA repair-targeting agents such as PARPi.
Somatic BRCA1/2 mutations have been identified in 4–6.4% of HGSCs, wherein they account for 14.2% of HRD cases [2,3]. Evidence suggests that the clinical benefit from PARPi in patients with somatic BRCA1/2-mutated HGSC is similar to that observed in those with germline BRCA1/2-mutated disease. In the phase III NOVA clinical trial, 19.7% (n=40) of BRCA1/2 mutations were classified as somatic [1]. The median progression-free survival (PFS) associated with niraparib as compared to that with placebo (20.9 vs. 11 months, hazard ratio [HR], 0.27; 95% confidence interval [CI], 0.08–0.9; P=0.02) in this subgroup was consistent with the value reported for patients with germline BRCA1/2-mutated disease (21 vs. 5.5 months, HR, 0.27; 95% CI, 0.17–0.41; P<0.001) [1].
BRCA1 promoter methylation has been identified as a potential biomarker of response to the PARPi rucaparib [4]. BRCA1-methylated tumors are negative for BRCA1 gene and protein expression, suggestive of a resultant HRD phenotype [5,6]. In addition, BRCA1-mutated and BRCA1-methylated OCs display similar gene signatures, as detected using gene expression and copy number analyses [7]. In the phase II open label ARIEL-2 study, 12/19 (63%) relapsed platinum-sensitive BRCA1-methylated OC patients responded to rucaparib as compared to an 80% response rate reported in BRCA1/2-mutated OC patients [4]. This early data suggest the potential role of BRCA1 methylation as a biomarker of response to PARPi.
Considering the benefit of PARPi in BRCA1/2 dysfunctional OC and the ongoing development of other agents targeting DNA repair, the knowledge of the prevalence and pattern of BRCA1/2 gene aberrations within an OC population is imperative. The use of tumor tissues offers the advantage of identifying additional potential somatic biomarkers of response to PARPi as compared to germline mutation testing alone. This information may serve as a guide to drug approval strategies for novel DNA repair targeting drugs at a national level, as the distribution of BRCA1/2 mutations varies between populations [8]. In Ireland, the frequency of BRCA1/2 gene aberrations in OC is yet to be examined. At the time of this study, genetic testing for BRCA1/2 mutations in OC in Ireland was carried out on the basis of clinical risk algorithms in a clinician-dependent manner.
Here, we sought to assess the BRCA1/2 gene profile in a cohort of Irish women with OC by determining the frequencies of BRCA1/2 mutations and BRCA1 methylation in tumors and their association with clinical characteristics and survival.
Go to : 
Materials and methods
1. Sample and data collection
We selected 111 patients with OC treated at 2 university teaching hospitals (including a national tertiary referral gynecologic oncology unit) between 2005 and 2013. All histological subtypes, stages, and grades were included to allow accurate assessment of BRCA1/2-mutated and BRCA1-methylated profiles. Borderline tumors were excluded. In total, 100 patients were retrospectively included from a prospective clinically annotated Discovary bioresource (St. James’s Hospital) after receiving ethical approval for this study (reference 2009/29/01). Patients provided written informed consents prior to specimen collection. Within this bioresource, all patients with epithelial OC with available and adequate tumor tissues (>30% neoplastic cell content [NCC]) were included. Eleven samples were obtained from the Beaumont Hospital Pathology Department after receiving approval from the hospital’s ethics committee (REC reference 12/02). Clinical data for these patients were retrospectively obtained through medical records. Patients recruited through both bioresources presented either to the outpatient department or as direct inpatient referrals. All survival data were updated to February 15, 2017. A pathologist specializing in gynecological cancers reviewed fresh-frozen paraffin-embedded (FFPE) tumor specimens for histology and NCC (as per the 2014 World Health Organization Classification). All specimens were obtained prior to chemotherapy (either at primary debulking surgery or peritoneal biopsy). Specimens with less than 30% NCC (n=20) were macrodissected prior to DNA extraction. The majority of the specimens had over 60% NCC.
2. Assessment of tumor BRCA1/2 defects
The DNA was extracted from FFPE tumor samples using the QIAamp DNA FFPE Tissue kit (Qiagen, Venlo, The Netherlands) according to the manufacturer’s protocol, and quantified using the dsDNA BR assay kit (Qubit, London, UK) as per the manufacturer’s instructions.
BRCA1 methylation status was assessed using the Methyl-Profiler DNA Methylation polymerase chain reaction (PCR) Array System (SABiosciences, Valencia, CA, USA) following the manufacturer’s protocol. In brief, DNA methylation-sensitive and methylation-dependent restriction enzymes were used to selectively digest non-methylated or methylated genomic DNA, respectively. After digestion, DNA samples were subjected to real-time PCR using primers flanking the regions of interest. The relative concentrations of differentially methylated DNA were determined by comparing the amount of each digest with that of a mock digest. A cutoff value of 10% methylation was used to define the methylation status of samples.
BRCA1 and BRCA2 genes were sequenced using the Tumor BRACAnalysis CDx assay (Myriad Genetics, Munich, Germany and Salt Lake City, UT, USA), as previously described [9]. Only deleterious or suspected deleterious mutations were included in analyses (as per the previously defined criteria [10]). Germline or somatic mutation status was not assessed, owing to the restrictions imposed by patients’ informed consent.
3. Statistical analysis
Statistical analysis was performed using SPSS® version 21.0 software. BRCA1/2 mutations and BRCA1 methylation were associated to the following variables: patient age, histology, International Federation of Gynecology and Obstetrics (FIGO) stage, degree of surgical cytoreduction, and platinum sensitivity using the Fisher’s exact test. Survival analyses were carried out for platinum-free interval (PFI), PFS, and overall survival (OS) to compare patients with BRCA1/2-mutated disease or BRCA1-methylated disease with patients carrying BRCA1/2 wild-type non-BRCA1-methylated (hereafter referred to as BRCA1/2-intact) tumors. PFI was defined as the interval between completion of chemotherapy and disease recurrence (as defined by the CA125/RECIST criteria), death, or date of last follow-up, whichever occurred first. PFS was defined as the interval between first surgical debulking or diagnostic biopsy (for patients receiving adjuvant or neoadjuvant chemotherapy, respectively) and disease recurrence (as defined by the CA125/RECIST criteria), death, or date of last follow-up, whichever occurred first. OS was defined as the interval between first surgical debulking or diagnostic biopsy (for patients receiving adjuvant or neoadjuvant chemotherapy, respectively) and death from any cause or date of last follow-up, whichever occurred first. All survival estimates were determined using Kaplan-Meier analysis (log-rank test). For all tests, a value of P<0.05 was considered statistically significant. Univariate and multivariate analyses of PFI, PFS, and OS were performed using the Cox proportional hazard regression model, which estimated HR and 95% CI.
Go to :
Results
1. Patient and disease characteristics
Patient and disease characteristics are listed in Table 1. Two patients were excluded from the analysis, one owing to insufficient tumor DNA and the other who carried both BRCA2 mutation and BRCA1 methylation, leaving a total cohort of 109 evaluable patients. The median age of patients at diagnosis was 59 years, and 63.3% (n=69) presented with advanced stage disease (FIGO stage III/IV). In total, 64.2% patients (n=70) had HGSC; stage III/IV HGSC comprised 53.2% (n=58) of the cohort, and 78.9% (n=86) and 5.5% (n=6) of patients received adjuvant and neo-adjuvant platinum-based therapy, respectively. None of the patients received PARPi therapy during the course of illness. The first PARPi therapy in Ireland was approved after the end of the follow-up period. Reasons for no primary chemotherapy included stage IA/IB disease (6%, n=7), peri-operative death (3%, n=3), age greater than 80 years old (3%, n=3), and other (3%, n=3). Microscopic surgical debulking (R0) was achieved in 66.2% (n=53/80) of patients with available data (data were missing for 26.6% [n=29] patients).
Table 1.
Patient and disease characteristics
| Parameter | No. of patients (%) |
|---|---|
| Age at diagnosis, median (range) | 59 (23–86) |
| FIGO stage | |
| I | 27 (24.8) |
| II | 13 (11.9) |
| III | 56 (51.4) |
| IV | 13 (11.9) |
| Pathology | |
| Serous | |
| High grade | 70 (64.2) |
| Low grade | 0 |
| Endometrioid | |
| Grade 3 | 3 (2.8) |
| Grade 2 | 11 (10.1) |
| Grade 1 | 3 (2.8) |
| Clear cell | 17 (15.6) |
| Mucinous, grade 1 | 3 (2.8) |
| Other | 2 (1.8) |
| Cytoreduction | |
| Microscopic | 53 (66.2) |
| 0–1 cm | 13 (16.3) |
| ≥1 cm | 14 (17.5) |
| Missing | 29 |
| Platinum sensitivity | |
| Resistanta) | 25 (23.1) |
| Partially sensitiveb) | 12 (11.1) |
| Sensitivec) | 55 (50.9) |
| No platinum chemotherapy | 16 (14.8) |
| Missing | 1 |
![]()
2. Frequency of BRCA1/2 aberrations
Methylation analysis revealed 10 tumors with at least 10% BRCA1 promoter methylation (median, 49.86%; range, 18.11–69.23%). All BRCA1-methylated tumors were stage III HGSC, totaling a BRCA1 methylation rate to 14.3% (n=10/70) in HGSC. Tumor BRCA1/2 gene sequencing revealed 18 pathogenic mutations (5 BRCA1 and 13 BRCA2) with an overall tumor BRCA1/2 mutation rate of 16.5% (n=18/109). No individuals were identified to carry more than a single tumor mutation, giving credence to each reported deleterious or suspected deleterious mutation. All mutations were identified in HGSC, and the combined germline and somatic BRCA1/2 mutation rate in HGSC was 25.7% (18/70). BRCA1 mutations were only observed in stage III/IV disease, while 3 BRCA2 mutations occurred in stage I/II cancers. In total, 16 of 18 mutations were classified as pathogenic as per the CLINVAR database [11], and 15 of 18 were curated as per the ENIGMA consortium [12]. BRCA1 mutations comprised one large genomic rearrangement, 2 frameshift, and 2 nonsense mutations, of which the c.1808C>A mutation has not been previously reported. The common BRCA1 Ashkenazi Jewish founder mutation c68_69del (also been reported as a separate British founder mutation [13,14]) was identified in one patient of unknown ethnicity. Most BRCA2 mutations were frameshift mutations except for a previously unreported nonsense mutation, c.19G>T, and one splice variant, c.631+1G>A, which were thought to result in abnormal mRNA splicing. Biochemical analysis revealed a similar mutation at this splice donor site that was found to be deleterious by Myriad Genetics laboratories. The BRCA2 mutation c.6486_6489del was identified in 4 samples from unrelated patients, thereby accounting for 31% of all BRCA2 mutations. This mutation has been previously reported as a germline variant in hereditary breast OC syndrome and observed in multiple ethnicities. Overall, 76.9% (10/13) of BRCA2 mutations were located in the RAD51-binding domain (exon 11), which is essential for homologous recombination DNA repair [15]. Three variants of unknown significance (2.7%) were identified in 2 patients (Table 2; Fig. 1 and [16]).
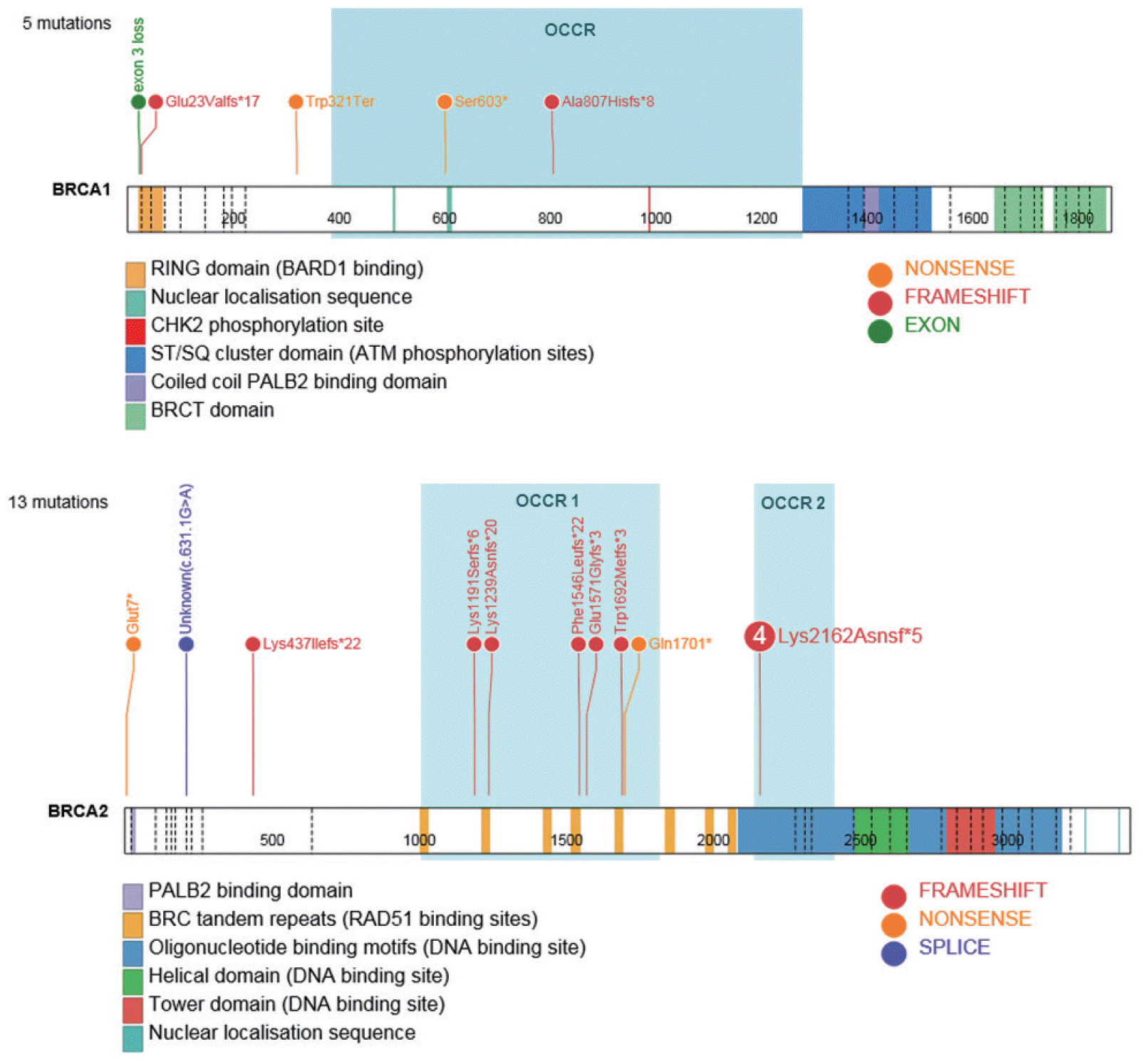 | Fig. 1.Localization of the identified BRCA1/2 mutations in BRCA1/2 proteins. Numbers on the protein graph correspond to amino acid locations; dashed lines delineate exons. Figure created using ProteinPaint software [16]. OCCR, ovarian cancer cluster region.
|
Table 2.
Details of BRCA1/2 mutations identified in the Irish cohort
![]()
3. Association of patient and disease characteristics with BRCA1/2 gene aberrations
Using the Fisher’s exact test, BRCA1/2 mutations were found to be significantly associated with stage III/IV disease (P=0.03) and HGSC (P<0.001). We failed to identify any association with younger age or platinum sensitivity. This is potentially owing to the small number and unknown germline/somatic status of BRCA1/2 mutations in our cohort. BRCA1 methylation also significantly differed between HGSC and non-HGSC (P=0.004). It was observed in 22.7% of HGSC but not detected among other OC subtypes, which comprised 34% of the entire cohort. Moreover, BRCA1-methylated OC was associated with FIGO stage III/IV disease (P=0.005). No significant correlation was identified between BRCA1 methylation and platinum sensitivity or other clinical variables (Table 3).
Table 3.
Correlation between tumour BRCA1/2 defects and clinico-pathological factors
![]()
4. Survival analyses
BRCA1/2 aberrations were identified in FIGO stage III/IV HGSCs, with the exception of 3 BRCA2 mutations (FIGO stage I/II disease). Survival analyses were restricted to FIGO stage III/IV HGSC (n=58) to minimize the bias of low stage and grade in the BRCA1/2 intact arm, thus allowing a more accurate assessment of the survival impact of BRCA1/2 aberrations. After a median follow-up of 3.8 (range, 0–11.5) years, patients with BRCA1/2-mutated tumors showed a trend toward improved PFI and PFS as compared to those with BRCA1/2-intact tumors (median survival: 17 vs. 6 months [HR, 0.48; 95% CI, 0.22–1.06; P=0.07] and 20 vs. 11 months [HR, 0.53; 95% CI, 0.26–1.09; P=0.08], respectively). The lack of expected statistical significance likely relates to the small sample size. OS significantly improved in patients with BRCA1/2-mutated tumors (median survival of 39 months) as compared to that in patients with BRCA1/2-intact disease (median survival of 26 months) (HR, 0.44; 95% CI, 0.19–0.99; P=0.045). No difference in survival was identified between the BRCA1-methylated and BRCA1/2-intact groups, as evident from the estimated median survivals of 5 vs. 6 months (HR, 1.15; 95% CI, 0.55–2.38; P=0.71), 10 vs. 11 months (HR, 0.99; 95% CI, 0.48–2.02; P=0.97), and 31 vs. 26 months (HR, 0.89; 95% CI, 0.40–1.95; P=0.76) for PFI, PFS, and OS, respectively (Fig. 2). After adjustment for residual disease in the multivariate analysis, BRCA1/2-mutated OC lost statistical significance with respect to improved OS (though the trend was similar), while the associations between BRCA1 methylation and PFI, PFS, and OS failed to show any significant change (Table 4).
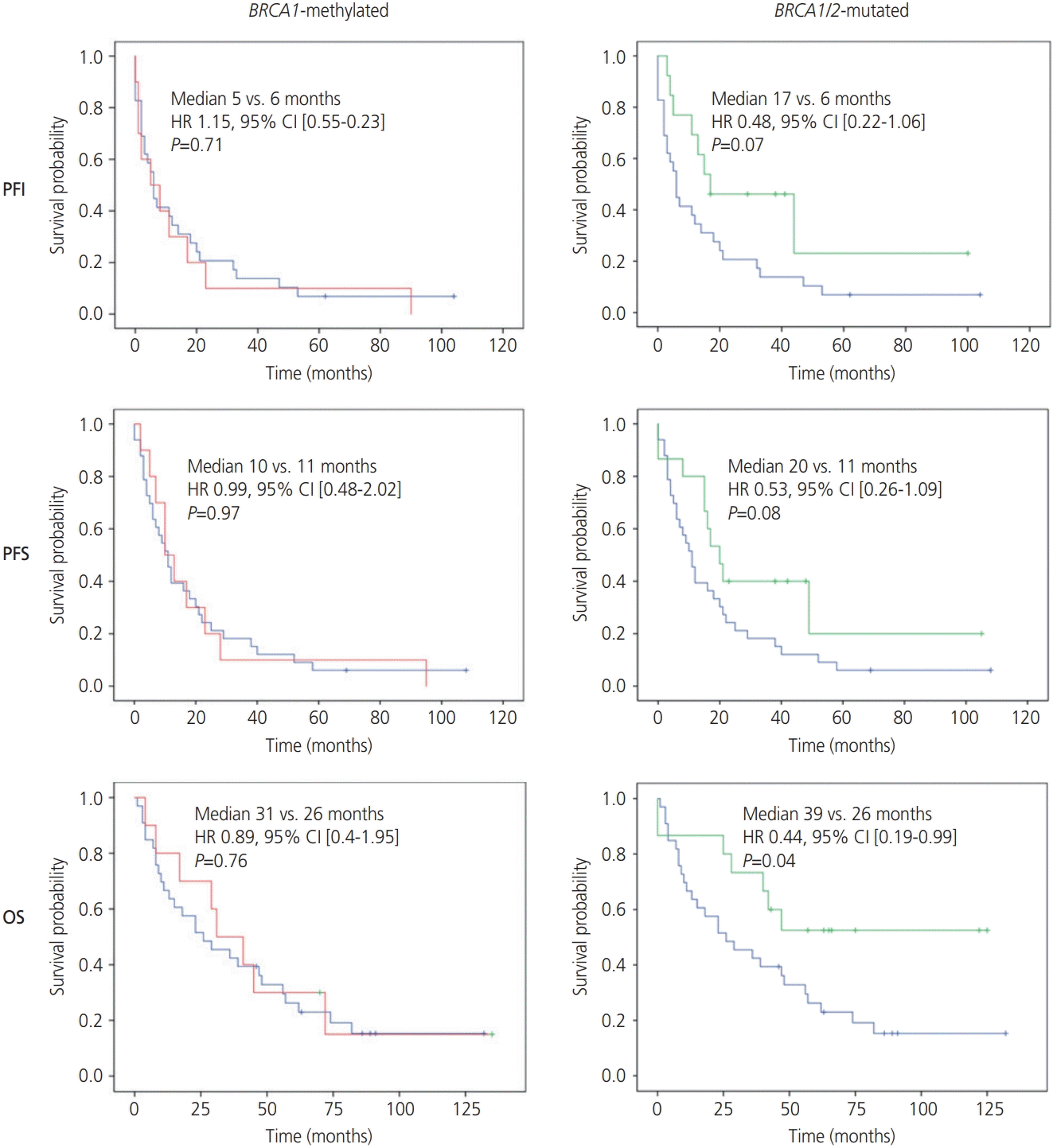 | Fig. 2.Survival analyses according to tumor-specific BRCA1/2 defect. Platinum-free interval (PFI), progression-free survival (PFS), and overall survival (OS) of patients with International Federation of Gynecology and Obstetrics (FIGO) stage III and IV high-grade serous tubal/ovarian cancer (HGSC). Comparison of patients with BRCA1-methylated HGSC and those with BRCA1/2-mutated HGSC to patients with BRCA1/2 wild-type non-BRCA1-methylated HGSC. In all graphs, blue curves indicate non-BRCA1-methylated; red curves indicate BRCA1-methylated; and green curves indicate BRCA1/2-mutated. HR, hazard ratio; CI, confidence interval.
|
Table 4.
Univariate and multivariate analyses for platinum-free interval (PFI), progression-free survival (PFS) and overall survival (OS) according to BRCA1/2 aberrations amongst advanced stage high grade serous ovarian cancers
![]()
Go to :
Discussion
This is the first study to assess the prevalence of BRCA1/2 aberrations in Irish patients with OC. We found an overall BRCA1/2 dysfunction rate of 25.7% (9.2% BRCA1-methylated and 16.5% BRCA1/2-mutated tumors). All cases were observed in HGSC, which comprised 64.2% of the study population. Within this subgroup, 14.3% of tumors were BRCA1-methylated and 25.7% were BRCA1/2-mutated, making an overall BRCA1/2 dysfunction rate of 40% in HGSC. Our findings are in line with those of other large studies, which reported germline/somatic BRCA1/2 mutation and BRCA1 methylation rates in the range of 19–27% and 10.5–14%, respectively, in HGSC [2,3,9,17]. Considering the therapeutic benefits of PARPi in BRCA1/2-mutated HGSC, and possibly in BRCA1-methylated HGSC [4], this degree of BRCA1/2 dysfunction within the most aggressive and lethal subtype of OC reinforces the crucial need outlined in the recent international guidelines to routinely test germline BRCA1/2 mutation status in patients with non-mucinous OC [18]. Testing FFPE tumor specimens for BRCA1/2 mutations using nextgeneration sequencing (NGS) may allow rapid analysis using low concentrations of DNA samples, making it a cost-effective approach. Tumor DNA sequencing differs from germline DNA sequencing owing to tumor heterogeneity and the risk of nucleic acid degradation during paraffin embedding process. As a result, concerns exist in using tumor BRCA1/2 mutation testing followed by germline testing of mutation-positive cases to comprehensively detect germline BRCA1/2 mutations. Our study was restricted in terms of testing the germline/somatic status of the identified mutations from tumor DNA. However, the tumor BRACAnalysis CDx test used in this study has been validated in different cohorts of HGSC FFPE specimens with matched blood samples. Upon application to FFPE specimens corresponding to each blood sample, this test correctly identifies all cases of germline-mutated BRCA1/2 HGSC in addition to 8.7% cases of somatic BRCA1/2 mutations [19]. Other reports using different BRCA1/2 panels and NGS platforms, where both germline and tumor tissues were available for analysis, have shown a discordance rate of ≤3% between tumor and blood-based testing for BRCA1/2 germline mutation [20]. Moreover, upfront tumor BRCA1/2 mutation followed by reflex germline mutation testing in mutation-positive patients may serve as a more cost-effective strategy than upfront germline mutation testing followed by subsequent tumor mutation testing in germline mutation-negative cases. Finally, the availability of tumor DNA allows BRCA1 methylation testing, further reinforcing the potential greater utility of tumor tissues in detecting therapeutic targets beyond germline BRCA1/2 mutations in a single test. However, further studies are warranted to determine the potential of BRCA1 methylation, in contrast to germline/somatic BRCA1/2 mutations, as a plausible therapeutic target.
In our study, BRCA1 methylation, like BRCA1/2 mutation, was associated with advanced stage HGSC. We failed to observe any association with younger age at diagnosis, contradicting the previous reports [17]. BRCA1 methylation decreased BRCA1 mRNA and protein expression in OC [5,6], suggestive of the sensitivity of HRD to platinum chemotherapy and PARPi. In vitro, BRCA1-methylated breast/OC cell lines demonstrate high sensitivity to cisplatin and olaparib as compared to BRCA1/2-intact cell lines [21,22]. We failed to translate these findings in the clinic, consistent with no survival difference between BRCA1-methylated OC and BRCA1/2-intact OC. Several large studies corroborate our observations [2,17], while others report a negative prognostic effect of BRCA1 methylation on survival [23]. Nevertheless, a few small studies have reported the superior platinum response and improved PFS amongst BRCA1-methylated tumors [24,25]. A larger study involving 213 patients with OC demonstrated similar values of HR for OS in germline BRCA1-mutated and BRCA1-methylated disease (HR, 0.88; 95% CI, 0.64–1.24 and HR, 0.89; 95% CI, 0.60–1.30, respectively; each group was compared to a BRCA1/2-intact population) [26]. Data regarding clinical responses of BRCA1-methylated OC to PARPi are limited to the ARIEL-2 study results, which reported a promising response rate of 63% in BRCA1-methylated tumors (n=12/19) [4]. These conflicting results are likely related to sample size and heterogeneity within BRCA1-methylated OC, as observed with BRCA1-mutated OC. While BRCA2-mutated OC consistently shows significant survival benefits, some reports revealed no survival difference between BRCA1-mutated OC and BRCA1/2 wildtype OC [27]. The survival benefit conferred by BRCA1 mutations may be potentially of lesser magnitude or diluted by the heterogeneous effect of different BRCA1 mutations [28] and mono- or biallelic BRCA1 mutations [29] on homologous recombination, thereby necessitating large cohorts to confirm this benefit [30]. Further, a significantly larger cohort of BRCA1-methylated OC would be necessary to detect survival benefits, if any.
The clinicopathological associations of BRCA1/2-mutated disease observed in the present study are similar to those previously reported. BRCA1/2 mutations were solely detected in HGSC, were associated with improved OS, and showed a trend toward significantly better PFI and PFS. The small sample size of our study limits the strength of survival analyses. A single BRCA2 mutation, c6486_6489del, accounted for 22% of all mutations detected. This is a known pathogenic germline mutation associated with hereditary breast OC syndrome. No tumor carried the BRCA1 c.2681_2682delAA variant, a founder mutation originating from Irish/West Scottish Celts [31]. We observed the predominance of BRCA2 mutations, with a BRCA2:BRCA1 mutation ratio of 2.6:1. In Caucasian HGSC cohorts, germline BRCA1 mutations were found to be 1 to 3 times more frequent than BRCA2 mutations [2,3,32]. BRCA1 mutations have higher penetrance and confer a 36–53% lifetime OC risk as compared to an estimated 11–25% lifetime risk with BRCA2 mutations [33]. The small sample size of our study may possibly lead to biased results. However, the heterozygote population distribution of BRCA1/2 mutations varies worldwide. An analysis of the Exome Aggregation Consortium and Exome Variant Server databases demonstrates a high frequency heterozygote BRCA2 germline mutations in some populations, with some populations having a very low rate of BRCA1 mutation carriers [8]. A very low frequency of BRCA1 heterozygote population could potentially explain our findings, though this cannot be verified in the absence of the frequency of BRCA1/2 mutation in Irish population. Two publications had reported a relatively higher BRCA2:BRCA1 mutation ratio [34,35], including a 2.7:1 ratio of BRCA2:BRCA1 mutations amongst 120 patients with non-mucinous OC undergoing routine BRCA1/2 germline mutation testing. Interestingly, the reports originate from Scotland/Northern Ireland and West Scotland. These populations share a common Celt genetic ancestry with the Irish population, thereby reinforcing the likelihood that our findings could be representative of the distribution of BRCA1/2 mutations amongst Irish patients with HGSC. Drawing such conclusions is however limited in our study by ethical constraints to perform germline testing on the identified tumor mutations to determine their germline/somatic status. As these reported mutations occurred singly in individuals and were classified as deleterious or suspected deleterious, a predominance of BRCA2 mutations has therapeutic and prognostic implications for patients because 76.9% of BRCA2 mutations identified were located in the RAD51-binding domain of the gene. These mutations are associated with improved PFS and OS in contrast to those located outside this domain [15].
In conclusion, we observed a BRCA1/2 dysfunction rate of 40% within the Irish HGSC population, owing to BRCA1/2 mutations and BRCA1 promoter methylation in tumors; we noted a unique predominance of BRCA2 over BRCA1 mutations. This observation reinforces the need for routine BRCA1/2 germline and somatic testing to facilitate therapy selection (PARPi and other forthcoming DNA repair targeting agents) and cancer prevention for germline mutation carriers and their relatives. A better understanding of the clinical and therapeutic relevance of BRCA1 methylation in OC is needed, given its potential to expand the therapeutic benefits of DNA repair targeted agents to a larger number of women with OC.
Go to :
 XML Download
XML Download