

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Glial cells play important roles in neuroprotection and maintaining homeostatic brain functions by secreting antioxidant enzymes, regulating synaptic function, and mediating immune defense against hazardous stimuli [1]. It is well known that Alzheimer's disease (AD) is strongly correlated with amyloid beta (Aβ) deposition. Aβ leads to the production of nitric oxide (NO) and pro-inflammatory cytokines in activated-glial cells, and the overproduction of inflammatory mediators and cytokines can cause neuronal cell death, contributing to AD progression [23]. One of the most critical functions of glial cells is the regulation of Aβ clearance and degradation. Previous studies have demonstrated that reduced activity of glial-mediated proteolytic enzymes, such as insulin degrading enzyme (IDE) and neprilysin (NEP), can promote Aβ accumulation [456]. In turn, Aβ-induced dysfunction of glial cells causes the downregulation of Aβ-degrading enzymes, and this decline in enzymatic activity is typically observed in the brain of AD patients [7]. Therefore, alleviating the release of pro-inflammatory cytokines and mediators and enhancing Aβ degradation activity may be potential therapeutic strategies for AD.
Paeonia lactiflora Pall. has been used as a traditional medical ingredient in Asian countries, such as Korea, China, and Japan [8]. Previous studies have shown that P. lactiflora exerts potential preventive and therapeutic effects in the treatment of neurodegenerative diseases, diabetes, cancer, and inflammation [9]. P. lactiflora contains more than 15 components including paeoniflorin (PF), albiflorin, lactiflorin, paeonin, and paeonol. Among them, a water-soluble monoterpene glycoside PF (molecular weight: 480.45) is the most abundant compound (> 90%) in P. lactiflora [1011]. It has been reported that P. lactiflora exerted beneficial health effects that are attributed to the anti-oxidant and anti-inflammatory properties of PF [1213]. In addition, PF has been shown to exhibit protective functions against neuronal apoptosis from Aβ25–35, N-methyl-D-aspartate, and glutamate [141516]. We previously showed that PF attenuated oxidative stress-induced SH-SY5Y neuronal cell death [17]. However, the role of PF on the Aβ-induced AD pathology in glial cells is not yet understood. Herein, we investigated the effect of PF on cell viability, NO production, and pro-inflammatory cytokine release in Aβ25–35-treated C6 glial cells. The mechanisms of PF underlying nuclear factor-kappa B (NF-κB)-mediated neuroinflammation and Aβ degradation were also examined.
Go to : 
MATERIALS AND METHODS
Instruments and reagents
PF (purity > 98%) was purchased from Cayman Chemical Co. (Ann Arbor, MI, USA). For cell culture, fetal bovine serum (FBS), Dulbecco's modified Eagle's medium (DMEM), and penicillin/streptomycin were obtained from Welgene (Daegu, Korea). Griess reagent, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), and dimethyl sulfoxide (DMSO) were supplied by Sigma Chemical Co. (St. Louis, MO, USA). Radioimmunoprecipitation assay (RIPA) buffer, 30% acrylamide bis solution, and pre-stained protein size markers were obtained from Bio-Rad (Hercules, CA, USA). Polyvinylidene fluoride (PVDF) membrane was obtained from Millipore (Billerica, MA, USA). Prior to use, PF was freshly prepared as a stock solution in DMSO.
Preparation of Aβ25–35
Aβ25–35 peptide (Sigma Chemical Co.) was dissolved in sterile distilled water to achieve a concentration of 1 mM. The Aβ25–35 stock solution was incubated to induce aggregation at 37°C for 3 days, and the solution was stored at −20°C until use. For cell experiments, the solution was diluted to 50 μM in the culture medium.
Cell culture
C6 glial cells were obtained from Korean Cell Line Bank (Seoul, Korea). The cells were grown in DMEM containing 1% penicillin/streptomycin and 10% FBS at 37°C in a humidified incubator containing 5% CO2. The cells were sub-cultured with 0.05% trypsin-EDTA in phosphate-buffered saline.
Cell viability
Cell viability was assessed by an MTT assay according to Mosmann's method [18]. C6 glial cells were harvested and plated in a 96-well plate at a density of 5 × 104 cells/mL. The cells were divided into the following groups: control (non-treated cells), Aβ (Aβ25–35-treated cells), PF1, PF5, and PF10 (Aβ25–35-treated cells pretreated with PF at 1, 5, and 10 μg/mL, respectively). The cells were treated with PF (1, 5, and 10 μg/mL) for 4 h, after which Aβ25–35 (50 μM) was added and the cells were further incubated for 24 h at 37°C in an incubator containing 5% CO2. After incubation, the cell supernatant was removed and MTT solution was added to each 96-well plate. The incorporated formazan crystals were solubilized with DMSO, and the absorbance of each well was read at 540 nm using a microplate spectrophotometer.
NO production
At confluence, the cells were plated in 96-well plates at a density of 5 × 104 cells/mL and cultured overnight. PF (1, 5, and 10 µg/mL) was added to the wells and incubated for 4 h, after which Aβ25–35 (50 μM) was added. After 24 h of incubation, NO production was determined using Griess reagent. To measure NO generation, the cell supernatant was mixed with Griess reagent at a 1:1 volume ratio in a 96-well plate, and the plate was incubated for 20 min at room temperature. The levels of NO production were measured at 540 nm using a microplate spectrophotometer [19].
Interleukin (IL)-6, IL-1β, and tumor necrosis factor-alpha (TNF-α) production
The supernatants of C6 glial cells treated with PF (1, 5, or 10 μg/mL) and Aβ25–35 (50 μM) were transferred to another plate. The levels of pro-inflammatory cytokine (IL-6, IL-1β, and TNF-α) production were measured using an ELISA kits (R&D Systems, Minneapolis, MN, USA) according to the manufacturer's instructions.
Western blot
Protein extracts were prepared from C6 glial cells according to the manufacturer's instructions using RIPA buffer supplemented with 1× protease inhibitor cocktail. Proteins were separated by electrophoresis in a precast 10%–13% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and blotted onto PVDF membranes. The membrane was blocked with 5% skim milk solution for 1 h at room temperature, and incubated overnight at 4°C with primary antibodies against the following proteins; inducible NO synthase (iNOS), cyclooxygenase-2 (COX-2), phospho NF-κB (pNF-κB), inhibitor kappa B-alpha (IκB-α), IDE (all from Santa Cruz biotechnology, Santa Cruz, CA, USA) and NEP (Millipore). The membrane was washed and incubated with appropriate horseradish peroxidase-conjugated secondary antibodies. Protein bands were visualized using a chemiluminescent imaging system (Davinci Chemi, Seoul, Korea) and the intensity of each protein expression was quantified using Image J software (National Institutes of Health, Bethesda, MD, USA). Protein expression levels were calculated by comparing the intensity of the bands with that of the control group.
Statistical analysis
The results are expressed as mean ± SD. Statistical significance was determined by one-way analysis of variance followed by Duncan's post hoc test using the SPSS program version 23 (SPSS Inc., Chicago, IL, USA). Significance was determined at P < 0.05.
Go to :
RESULTS
Effect of PF on the viability in Aβ25–35-induced C6 glial cells
Our preliminary study showed that treatment with PF at a range of concentrations from 0.1 to 10 µg/mL did not affect the condition in C6 glial cells. However, PF at 25 µg/mL significantly decreased cell viability compared to that of the control group, indicating that PF exerts cytotoxic effects on C6 glial cells at 25 µg/mL (data not shown). Based on these findings, we further assessed the effect of PF on Aβ25–35-treated C6 glial cells at a concentration range of from 1 to 10 µg/mL. As illustrated in Fig. 1, Aβ25–35 significantly diminished cell viability (39.95%), compared with that of the non-treated group (100%), whereas PF increased the cell viability compared with only Aβ25–35-treated group. Especially, PF at 10 μg/mL significantly restored cell survival to 45.82%. These results indicate that PF attenuated Aβ25–35-induced damage to C6 glial cells.
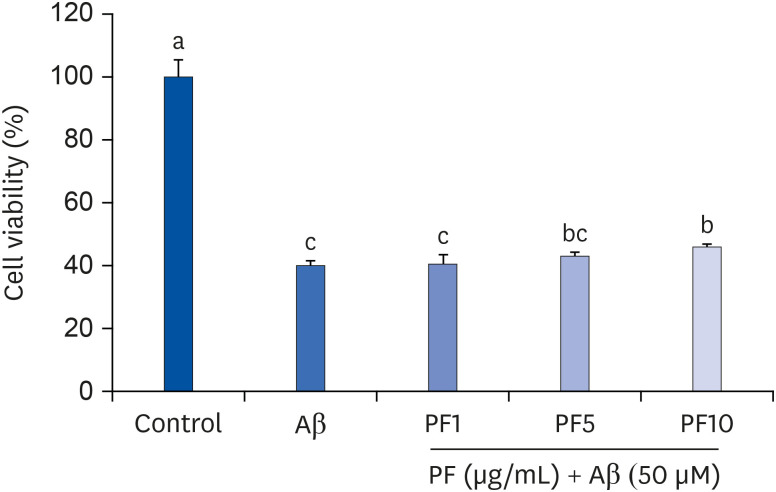 | Fig. 1Effect of PF on the viability in Aβ25–35-treated C6 glial cells. ‘Control’ represents non-treated cells, ‘Aβ’ represents Aβ25–35-treated cells, ‘PF1’, ‘PF5’, or ‘PF10’ represent the 3 concentrations of PF treatment (1, 5, and 10 μg/mL, respectively) in Aβ25–35-treated cells. Values are mean ± SD.Aβ, amyloid beta; PF, paeoniflorin.
a-cMeans with different letters are significantly different (P < 0.05) as determined by Duncan's multiple range test.
|
Effect of PF on NO production in Aβ25–35-induced C6 glial cells
We then examined whether PF regulates NO production in Aβ25–35-induced C6 glial cells. The NO levels were measured using Griess reagent. We found that C6 glial cells treated with Aβ25–35 significantly increased NO levels (100%), compared with control group (83.57%) (Fig. 2). Meanwhile, pre-treatment with PF suppressed the NO production in Aβ25–35-induced C6 glial cells. In particular, PF 10 μg/mL significantly decreased the levels of NO generation, showing 84.29%.
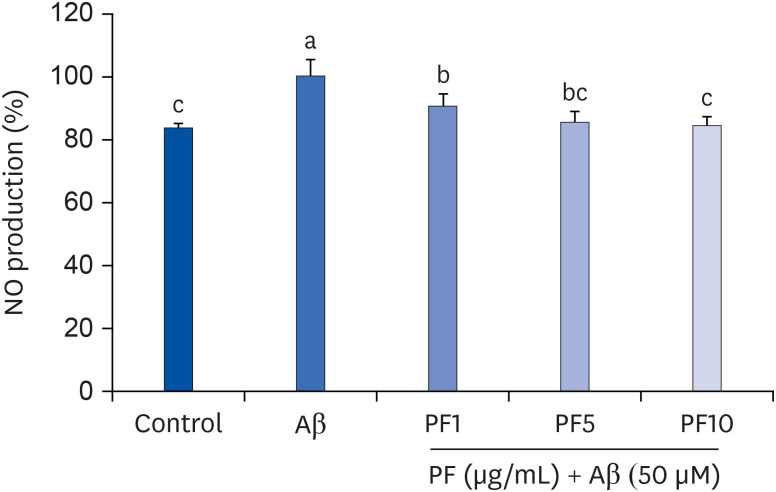 | Fig. 2Effect of PF on NO production in Aβ25–35-treated C6 glial cells. ‘Control’ represents non-treated cells, ‘Aβ’ represents Aβ25–35-treated cells, ‘PF1’, ‘PF5’, or ‘PF10’ represent the 3 concentrations of paeoniflorin treatment (1, 5, and 10 μg/mL, respectively) in Aβ25–35-treated cells. Values are mean ± SD.NO, nitric oxide; Aβ, amyloid beta; PF, paeoniflorin.
a-cMeans with different letters are significantly different (P < 0.05) as determined by Duncan's multiple range test.
|
Effect of PF on the production of pro-inflammatory cytokines in Aβ25–35-induced C6 glial cells
We further investigated whether PF inhibits the production of pro-inflammatory cytokines in Aβ25–35-induced C6 glial cells. As shown in Fig. 3, Aβ25–35 treatment led to a significant increase in IL-6, IL-1β, and TNF-α generation in C6 glial cells. In contrast, PF significantly attenuated IL-6, IL-1β, and TNF-α production in Aβ25–35-treated C6 glial cells. In particular, IL-6 and TNF-α production were markedly inhibited by PF 10 μg/mL to levels similar to those of control group. These results suggest that PF suppressed the release of Aβ25–35-induced pro-inflammatory cytokines in C6 glial cells.
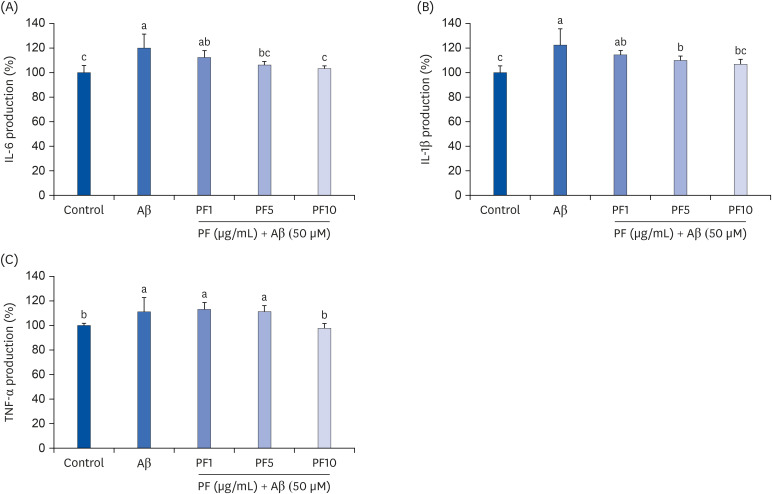 | Fig. 3Effect of PF on the production of IL-6 (A), IL-1β (B), and TNF-α (C) in Aβ25–35-treated C6 glial cells. ‘Control’ represents non-treated cells, ‘Aβ’ represents Aβ25–35-treated cells, ‘PF1’, ‘PF5’, or ‘PF10’ represent the 3 concentrations of paeoniflorin treatment (1, 5, and 10 μg/mL, respectively) in Aβ25–35-treated cells. Values are mean ± SD.IL, interleukin; Aβ, amyloid beta; PF, paeoniflorin; TNF-α, tumor necrosis factor-alpha.
a-cMeans with different letters are significantly different (P < 0.05) as determined by Duncan's multiple range test.
|
Effect of PF on the protein expression of iNOS, COX-2, pNF-κB, and IκB-α in Aβ25–35-induced C6 glial cells
We examined whether the NF-κB signaling pathway was involved in mediating the anti-inflammatory property of PF in Aβ25–35-induced inflammatory conditions. Our results revealed that Aβ25–35 treatment upregulated the protein expression of iNOS and COX-2 (Fig. 4). Conversely, PF downregulated the protein expression levels of iNOS and COX-2, suggesting that PF blocked NO and pro-inflammatory cytokine production by inhibiting iNOS and COX-2. In terms of the regulation of iNOS and COX-2 expression, we investigated the effect of PF on the activity of the transcription factors, NF-κB and IκB-α. As shown in Fig. 4, the level of pNF-κB was significantly upregulated by treatment with Aβ25–35, compared with control group. However, PF attenuated the Aβ25–35-induced protein expression of pNF-κB, and which was linked to an increase in IκB-α expression. This finding suggests that the anti-inflammatory effect of PF may be associated with the inhibition of transcriptional activity of NF-κB in Aβ25–35-treated C6 glial cells.
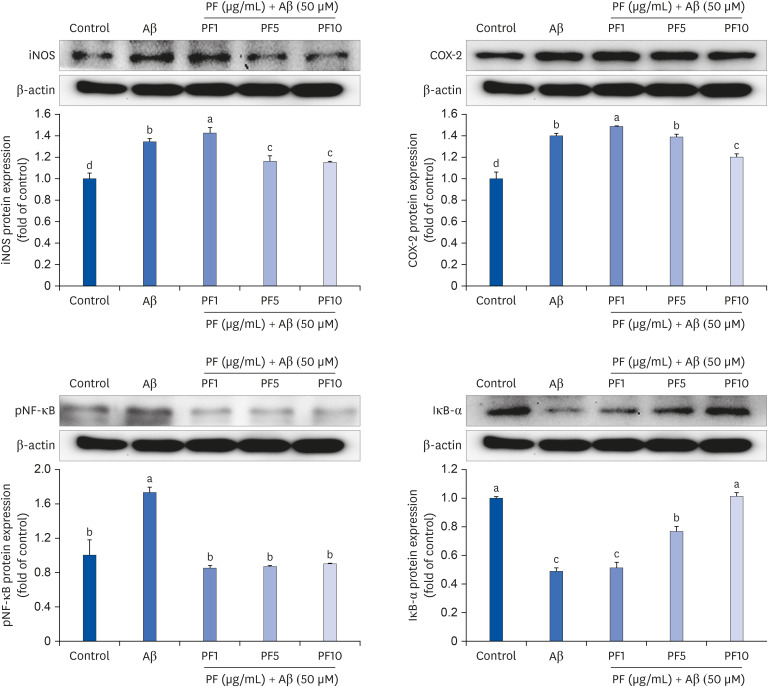 | Fig. 4Effect of PF on the protein expression of iNOS, COX-2, pNF-κB, and IκB-α in Aβ25–35-treated C6 glial cells. ‘Control’ represents non-treated cells, ‘Aβ’ represents Aβ25–35-treated cells, ‘PF1’, ‘PF5’, or ‘PF10’ represent the 3 concentrations of paeoniflorin treatment (1, 5, and 10 μg/mL, respectively) in Aβ25–35-treated cells. Values are mean ± SD.iNOS, inducible NO synthase; Aβ, amyloid beta; PF, paeoniflorin; COX-2, cyclooxygenase-2; pNF-κB, phospho nuclear factor-kappa B; IκB-α, inhibitor kappa B-alpha.
a-dMeans with different letters are significantly different (P < 0.05) as determined by Duncan's multiple range test.
|
Effect of PF on the protein expression of IDE and NEP in Aβ25–35-induced C6 glial cells
To analyze the effect of PF on Aβ protease activity in Aβ25–35-treated C6 glial cells, the protein expression of Aβ-degrading enzymes IDE and NEP was assessed. Compared with the control group, Aβ-stimulated cells showed reduced protein expression of IDE and NEP, which was conversely upregulated by 10 μg/mL of PF (Fig. 5). Our findings suggest that PF leads to the proteolytic activity of Aβ, possibly contributing to the alleviation of Aβ-induced neuroinflammation.
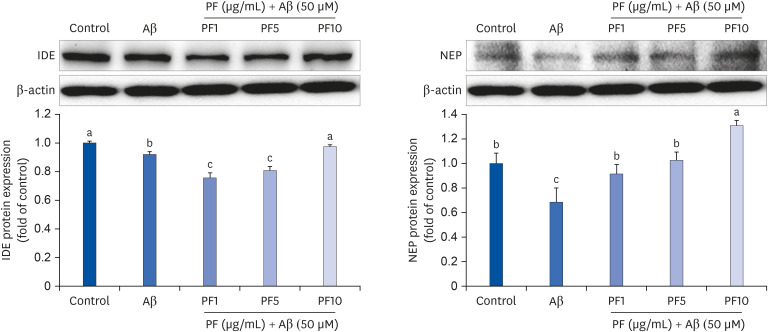 | Fig. 5Effect of PF on the protein expression of IDE and NEP in Aβ25–35-treated C6 glial cells. ‘Control’ represents non-treated cells, ‘Aβ’ represents Aβ25–35-treated cells, ‘PF1’, ‘PF5’, or ‘PF10’ represent the 3 concentrations of paeoniflorin treatment (1, 5, and 10 μg/mL, respectively) in Aβ25–35-treated cells. Values are mean ± SD.IDE, insulin degrading enzyme; Aβ, amyloid beta; PF, paeoniflorin; NEP, neprilysin.
a-cMeans with different letters are significantly different (P < 0.05) as determined by Duncan's multiple range test.
|
Go to :
DISCUSSION
Aβ-mediated neuroinflammation is an important mechanism that contributes to AD progression [20]. Glial cells are immune cells in the brain that represent the initial defense against Aβ accumulation. Several studies have reported that glial cells, which play critical roles as regulator of Aβ levels in the brain, are involved in enzymatic degradation and phagocytosis [721]. The upregulation of degrading enzymes, such as IDE and NEP, effectively promotes Aβ removal in the brain. On the other hand, the overproduction of pro-inflammatory cytokines and mediators by Aβ can reduce the activity of these Aβ-degrading enzymes in glial cells, resulting in neuronal degeneration. Thus, inhibition of this vicious cycle seems to be a promising therapeutic strategy for AD.
Various Aβ fragments, such as Aβ1–40, Aβ1–42, and Aβ25–35, have been reported to aggregate and exert neurotoxic effects in AD [22]. Aβ25–35 is the shortest fragment that has been shown to retain the physical and biological properties of full-length Aβ [23]. Among the fragments, Aβ25–35 is low-molecular-weight oligomer that is easily soluble. Thus, it is widely used in AD-relevant in vitro and in vivo studies instead of the endogenous Aβ1–40 or Aβ1–42 fragment [24]. Consistent with this evidence, many studies have shown that Aβ25–35 induced toxicity and inflammatory damage by inducing apoptosis and morphological abnormalities in glial cells [252627]. Therefore, we used Aβ25–35 for this study of the protective effect of PF predicted to modulate Aβ toxicity.
In the present study, Aβ25–35 significantly reduced the viability of C6 glial cells, whereas pretreatment with PF prevented the decrease in cell viability. These results suggest that PF exerted neuroprotective effects against Aβ-induced toxicity in C6 glial cells. NO has been reported as a critical role in variety of physiological responses, such as neurotransmitter, immune regulator, mediator of vascular relaxation [28]. However, high levels of NO can lead to the generation of other reactive nitrogen species such as peroxynitrite, which is a toxic molecule involved in inflammation. In addition, increased expression of iNOS in glial cells was shown to elevate NO concentration, leading to neuronal cell death [29]. Therefore, preventing NO overproduction has been considered as a therapeutic strategy in neurodegenerative disorders, such as AD [3031]. Aβ is a potent stimulator of inflammatory molecules in a variety of cell types. We previously demonstrated that treatment of Aβ25–35 induced the production of reactive oxygen species (ROS) and NO in C6 glial cells, indicating that Aβ25–35 is capable of inducing oxidative stress and inflammatory response [32]. As reported previously, our present results also showed that Aβ25–35 prominently increased NO production compared with that in non-treated cells. However, PF significantly suppressed the excessive production of NO in Aβ25–35-induced C6 glial cells.
The release of pro-inflammatory cytokines in glial cells is one of the primary responses leading to the inflammatory cascade, resulting in subsequent neurodegeneration [33]. In glial cells, Aβ stimulation leads to the secretion of inflammatory cytokines, such as prostaglandin E2, IL-1β, IL-6, and TNF-α [3435]. These cytokines are implicated in the enhanced enzymatic activity of iNOS and COX-2 through the transcriptional pathway, resulting in the induction of neuroinflammation. Consistent with previous findings, the results of the present study indicated that the levels of IL-6, IL-1β, and TNF-α were significantly elevated by Aβ25–35 in C6 glial cells. However, IL-6, IL-1β, and TNF-α production were significantly suppressed by PF in Aβ25–35-treated cells. We also observed that Aβ25–35-induced upregulation of iNOS and COX-2 protein expression was reduced by treatment of PF in C6 glial cells. PF was previously shown to significantly decreased the excessive release of NO, IL-1β, and TNF-α in lipopolysaccharide (LPS)-stimulated hippocampal cells and primary microglial cells [36]. In addition, the release of pro-inflammatory mediators (IL-1β, TNF-α, iNOS, and COX-2) was inhibited by PF in the brain of ischemia-injured rats [37]. We showed that Aβ-stimulated NO production was prevented by treatment with PF at 1, 5, and 10 μg/mL. However, pretreatment with 1 μg/mL PF did not have a notable influence on the protein expression of iNOS and COX-2 in Aβ25–35-induced C6 glial cells. The cause of this result is unclear, but could perhaps be related to iNOS-independent mechanisms. It has been reported that NO is produced in glial cells not only by iNOS but also by other isoforms of NOS; neuronal NOS (nNOS) and endothelial NOS (eNOS) [38]. In addition, it was suggested that NO suppression is not always mediated by COX-2 downregulation [3940]. Although the precise mechanism by which 1 μg/mL PF inhibited NO production under inflammatory stimuli is yet to be elucidated, we speculate that at this concentration, PF may trigger iNOS-independent mechanisms such as the generation of superoxides and/or peroxynitrite from eNOS and nNOS activity, which may contribute to the inhibition of NO production.
Regulation of the NF-κB signaling pathway is strongly implicated in the treatment and prevention of AD. Activated NF-κB has been observed in neuronal and glial cells in regions surrounding the Aβ plaque, leading to accelerated AD symptoms with neuroinflammation and further Aβ accumulation by amyloidogenesis [41]. Following NF-κB activation, IκB proteins are phosphorylated and degraded. According to previous research, Aβ stimulates the activation of the transcription factor NF-κB, which enhances pro-inflammatory cytokine production [3442]. To evaluate the underlying mechanism of PF on the production of inflammatory cytokines and mediators, we analyzed the protein expression of pNF-κB and IκB-α. We revealed that Aβ-dependent nuclear translocation of NF-κB and IκB-α degradation were markedly inhibited by treatment of PF in C6 glial cells. These results support that PF seemed to inhibit NF-κB activation through a regulatory mechanism involving the generation of the cleaved form of IκB-α. We further propose that the inhibition of Aβ-induced NO and pro-inflammatory cytokine production is in accordance with inactivation of NF-κB-dependent signaling pathway.
Aβ can be degraded by glial proteases such as IDE and NEP. Interestingly, decreased levels of IDE and NEP have been observed in the brains of AD patients, indicating that a reduction in Aβ degradation may contribute to AD progression [43]. In vivo studies have demonstrated that upregulation of IDE and NEP expression prevented AD pathology by removing Aβ plaques [44]. However, Aβ and amyloid precursor protein levels were markedly increased in the brain of an IDE-knockout animals [45]. To investigate the relationship between the protective role of PF against Aβ-mediated neuroinflammation and Aβ degradation, we examined the protein expression of IDE and NEP in Aβ25–35-treated C6 glial cells by Western blot. Our results indicated that the protein levels of IDE and NEP were significantly decreased by Aβ25–35. In contrast, PF significantly increased the levels of these proteins, compared with those in Aβ25–35–treated cells. Our findings suggest that PF may promote Aβ degradation by regulating the activity of Aβ-degrading enzymes, and these effects contribute to attenuation of Aβ-induced inflammatory responses in glial cells.
Accumulating evidence has demonstrated that PF exerts protective effects against cognitive dysfunction by modulating neuroinflammation and Aβ deposits in vivo. PF administration ameliorated cognitive deficit, which was related to the inhibition of NF-κB signaling pathway [4647]. Zhang et al. [48] suggested that PF treatment significantly reduced Aβ plaque deposition in the brain of APP/PS1 transgenic mice. In addition, a recent study showed that PF administration via intraperitoneal injection for 28 days significantly decreased the Aβ plaque burden in the cortical and hippocampal areas of 5WFAD mice, indicating that the neuroprotective effect of PF was attributed to reduced Aβ accumulation [49]. The anti-inflammatory role of PF in the brain has also been reported in different cell types in the central nervous system. In SH-SY5Y neuronal cells, Aβ25–35-induced ROS production and mitochondrial apoptotic pathway were inhibited by treatment with PF [14]. Liu et al. [50] demonstrated that PF pretreatment prevented the excessive production of pro-inflammatory mediators and chemotactic factors by regulating NF-κB signaling pathway and the vascular endothelial growth factor (VEGF)/VEGF receptor 1 axis in Aβ1–42-treated primary microglial cells from Sprague Dawley rats. Furthermore, PF significantly blocked LPS-stimulated neurotoxicity and inflammatory responses in primary microglial cells [36]. PF could quickly penetrate the blood-brain-barrier to reach the brain region and maintain a high concentration, supporting the protective effect of PF on glial cells [51]. Although the anti-inflammatory role of PF linked to the reduction in Aβ formation and regulation of the amyloidogenic pathway is still unknown in Aβ-induced glial cells, PF may play a beneficial role in reducing Aβ-induced neuroinflammation by promoting Aβ clearance and enhancing the activity of degradation enzymes in the brain.
Taken together, PF appears to act as a potential inhibitor of Aβ-induced NF-κB activation by downregulating iNOS and COX-2 protein expression and inhibiting the production of NO and pro-inflammatory cytokine. Furthermore, the protein levels of IDE and NEP were significantly enhanced by treatment with PF, indicating that PF modulated inflammatory response by promoting Aβ degradation. Although further studies are required to investigate whether PF reduces Aβ accumulation in the brain of animal models, PF has shown protective properties against AD by attenuating neuroinflammation and facilitating Aβ degradation. These findings suggest that PF could be potential therapeutic agent for the prevention of Aβ-induced neurodegenerative diseases.
Go to :
 XML Download
XML Download