

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Chromosome 4q deletion syndrome is a rare disease caused by interstitial or terminal deletion of the long arm of chromosome 4 [1-3]. These deletions are typically de novo, although some cases have been found to result from an unbalanced product of parental reciprocal translocation [4]. The majority of 4q deletion cases are associated with a 2–15.1 Mb deletion of the q11–q31 region, with substantial phenotypic variation. However, only a small number of previously reported cases have been confirmed by high-resolution array comparative genomic hybridization (aCGH).
In the present study, we report the case of a patient with an interstitial deletion of a maximum, 22.8 Mb on chromosome 4q. Clinical and genetic characteristics were compared against previously reported patients with deletion of the same region, confirmed by aCGH. Further, we discuss candidate genes, associated with this syndrome, as potential markers, which could provide new insights into the phenotypic spectrum and genotype-phenotype correlation.
Go to : 
CASE REPORT
The present case looks at an 18-year-old male patient with an unremarkable family history. He is the third child of non-consanguineous, healthy, Korean parents and was born at a gestational age of 40 weeks, at 2.8 kg, by vaginal delivery. Upon presentation, his height and weight were less than 1 percentile. The patient fur-ther presented with microcephaly and clear dysmorphic facial features, including cranial asymmetry, prominent forehead protrusion, a bilateral epicanthal fold, hypertelorism, depressed nasal root with a broad or beaked nose and low-set, simple ears with preauricular dimples. No spots or piebaldism were observed. A neurological examination indicated hypotonia, but the deep tendon reflex was well preserved. Exophthalmos, ptosis, and strabismus were observed in both eyes with a cataract also present in the left eye. A dental examination revealed white plaque, due to the generalized amelogenesis imperfecta, with normal maxillomandibular growth, but bimaxillary protrusion was evident. Due to substantially delayed bone maturation, bone age was delayed (Fig. 1A). Abnormalities in the spine (Fig. 1B) and both feet (Fig. 1C) were also present. There were no abnormalities in the right hip, according to both hip radiographs; however, sequelae of Legg-Calvé-Perthes was observed in the left hip, with a short, broad left femoral head and relative shortening of the left limb with coxa magna deformity (Fig. 1D). This caused a discrepancy of 2 cm in leg length.
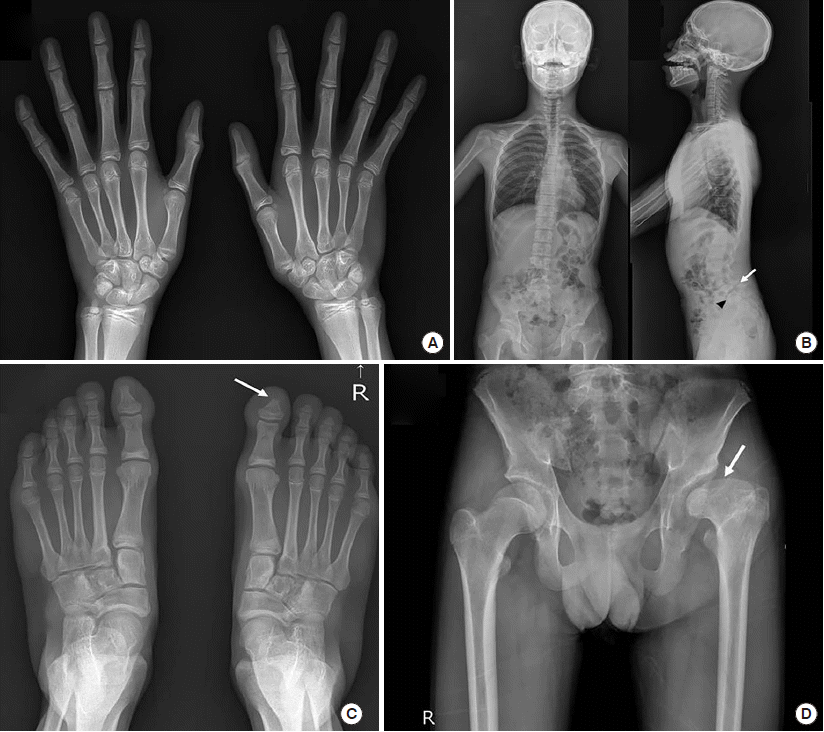 | Fig. 1Skeletal radiographs of the proband. (A) Both AP hand radiographs showed no fusion of the phalangeal physis’ and the bone age was considered to be in the order of 14 years, determined by the Greulich-Pyle method at the age of 17.3 years. (B) Whole spine, AP and lateral, radiographs showed minimal scoliotic curvature of the T-L spine, loss of normal lordotic curvature of the cervical spine, moderate spondylolytic spondylolisthesis at L5 on S1 (arrow), and a deformity of the anterosuperior corner of the S1 body (arrowhead). (C) AP radiographs of both feet showed deformity of the right first distal phalangeal bone (arrow). (D) Both hip AP radiographs showed sequelae of Legg-Calve-Perthes disease in the left hip, with a short, broad femoral head (arrow) and relative shortening of the left limb, in keeping with a coxa magna deformity.
|
The patient had a no history of seizure but did present with a learning disability and speech disorder, along with severe developmental delay. A psychiatric evaluation revealed signs of attention deficit hyperactivity disorder (ADHD) and autism spectrum disorder (ASD). An investigation of his pubertal status showed a testes volume of 18/18 mL, pubic hair score of 4–5, genitalia score of 5 with appropriate adult external genitalia overall. His serum testosterone, luteinizing hormone and follicle stimulating hormone levels were all elevated, at 3.95 (reference range: 1.7–8.6 mIU/mL), 3.37 (1.5–12.4 mIU/mL) and 8.57 (1.88–8.82 ng/mL), respectively. No abnormal findings were observed in complete blood cell count, urinalysis, biochemical tests, hearing tests, electroencephalography, echocardiography, pelvic sonography, and the kidney-uretersbladder sonography survey.
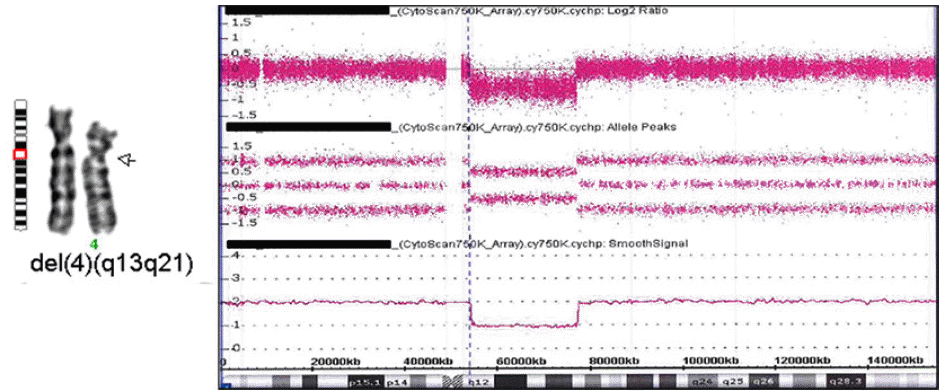
Cytogenetic analysis and aCGH of the patient revealed an interstitial, heterozygous deletion of chromosome 4q: 46,XY,del(4)(q12q21.1) arr[GRCh38] 4q12q21.1(53575744_76379310)x1 (Fig. 2) with the maximum length of the deleted region being 22.8 Mb. His mother had a normal karyotype, but no chromosome analysis was performed on his father. Neither parent showed any of the phenotypes of the proband. Chromosome analysis of peripheral blood lymphocytes was performed, according to standard techniques, at a 550-band level. The aCGH was performed using the Affymetrix Cytoscan 750K array. Copy number variants were compared with public databases, including the DECIPHER database (http://decipher.sanger.ac.uk) and USCS Genome Browser (http://www.genome.ucsc.edu/cgi-bin/hgGateway). No pathogenic or likely pathogenic variant was found in whole-exome sequencing using the peripheral blood sample of the proband. All exon regions of all human genes (~22,000) were captured using Agilent’s SureSelect kit and the captured regions of the genome were sequenced with the Illumina sequencing platform.
The deleted region of the proband comprised 109 known Mendelian Inheritance in Man (MIM) genes, including 25 genes associated with disease phenotypes; however, few of these genes were associated with specific phenotypes (Fig. 3A). The patient and his parents provided written informed consent for the publication of patient information.
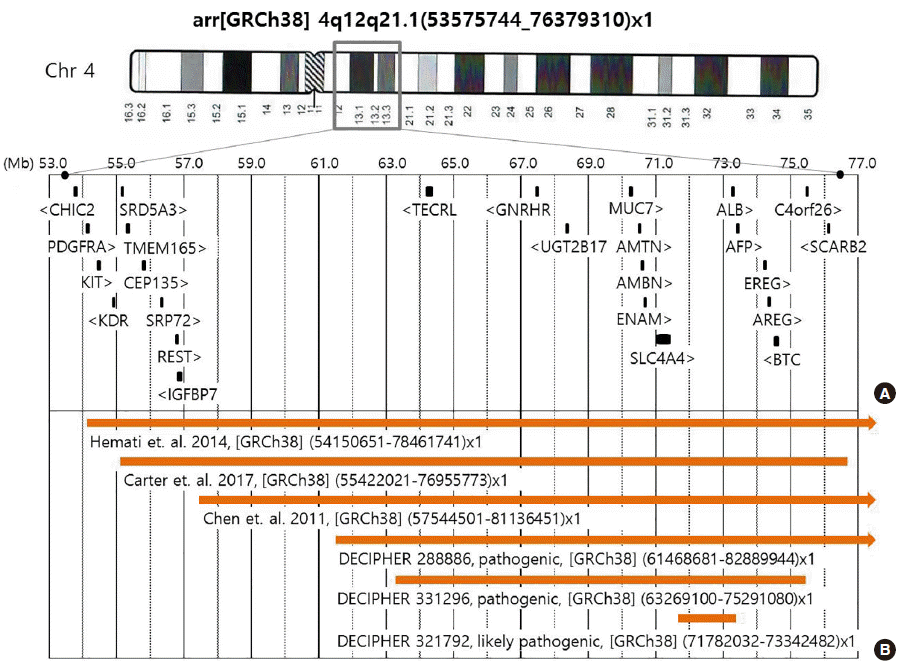 | Fig. 3The deleted region of the proband and diagrammatic comparison of previously reported cases. (A) Significant MIM genes in the region of the 22.8-Mb deletion were detected by whole-genome SNP array in the proband. (B) The regions of previously published cases, with sufficient clinical data and confirmed by aCGH at the same loci, and DECIPHER cases of pathogenic and likely pathogenic deletions, within the deleted region of the proband.
|
Go to :
DISCUSSION
Many patients with 4q proximal deletion of various sizes have been reported to date, including 4q12q21.1. Capalbo et al. [5] summarized the clinical characteristics of 31 patients with pure 4q proximal deletions; however, detailed information on the deleted region was not provided for these patients. Only three of these cases had similar sized deletions (21.59–24.37 Mb) at the same loci as our present case, and this was confirmed by aCGH (Fig. 3B). In addition, the DECIPHER database (version 9.23, released on May 23, 2018) showed three cases recorded as “pathogenic” or “likely pathogenic”, which were smaller sized deletions of this region, as pure copy number variations (DECIPHER number 288886, 321792, and 331296) (Fig. 3B). Therefore, including the present case, there are only seven cases of 4q proximal deletion confirmed with aCGH and detailed clinical information (Table 1). The common characteristics of these cases include mental retardation, learning disability, growth retardation, minor facial anomalies, hypotonia, skeletal abnormalities, and eye anomalies. Therefore, more in-depth comparisons of these cases could prove useful in understanding the genotype-phenotype relationship of the proximal 4q region.
Table 1
The clinical features of the proband along with other previously reported cases
| Our case | Carter et al., 2017 [9] | Hemati et al., 2015 [8] | Chen et al., 2011 [10] | Decipher 288886 | Decipher 331296 | Decipher 321792 | |
|---|---|---|---|---|---|---|---|
| Deleted region [GRCh38] | 53,575,744-76,379,310 | 55,422,021-76,955,773 | 54,150,651-78,461,741 | 57,544,501-81,136,451 | 61,468,681-82,889,944 | 63,269,100-75,291,080 | 71,782,032-73,342,482 |
| Deleted size | 22.80 Mb | 21.53 Mb | 24.31 Mb | 23.59 Mb | 21.42 Mb | 12.02 Mb | 1.56 Mb |
| Karyotype/chromosome locus | 46,XY,del(4)(q12q21.1) | 46,XY,del(4)(q12q21.1)/47,idem,+r(4) | 46,XX,del(4)(q12q21.21) | 46,XX,del(4)(q12q21.2) | 4q13.1-q21.22* | 4q13.1-q13.3* | 4q13.3-q13.3* |
| Gender/age | M/17 yr | M/6 mon, 22 mon | F/7 yr 3 | F/13 6 mon | ?/? | M/? | F/? |
| Inheritance | unknown | de novo | unknown | de novo | unknown | de novo | maternal |
| Intellectual/learning disability | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Growth delay | Yes | Yes | Yes | Yes | ND | ND | ND |
| Short stature | Yes | Yes | Yes | Yes | ND | ND | Yes |
| Facial dysmorphism | Yes | Yes | Yes | Yes | ND | ND | ND |
| Microcephaly | Yes | ND | 10 percentile at 18 mon, 25-50 percentile later | Yes | ND | ND | Yes |
| Eye anomalies | Yes | ND | Yes | Yes | ND | ND | ND |
| Amelogenesis defect | Yes | ND | No | Yes | ND | ND | ND |
| Piebaldism | No | Yes | No | No | ND | ND | ND |
| Hypotonia | Yes | ND | Yes | Yes | ND | ND | ND |
| Abnormality of the skeletal system | Yes | ND | Yes | ND | ND | ND | Yes |
| Other | ADHD,Autism | hepatomegaly | Hyperextensible skin, hypermobile joints | Delayed puberty | Puberty and gonadal disorders |
![]()
As shown in Table 1, mental retardation and/or learning disability was a common clinical finding in all seven cases (100%). The chromosomal region currently analyzed (4q) was found to be associated with two distinct syndromes linked to mental retardation and growth retardation: Kahrizi syndrome (MIM#612713) and renal tubular acidosis, proximal, with ocular abnormalities and mental retardation (RTA-OA-MR, MIM#604278). Kahrizi syndrome, which can be caused by a homozygous mutation of SRD5A3 (MIM* 611715) [6], is an autosomal recessive, neurodevelopmental disorder characterized by mental retardation, cataracts, coloboma, kyphosis, and coarse facial features. RTA-OA-MR is also an autosomal recessive disease, and it is caused by a homozygous mutation in SLC4A4 (MIM*603345) [7]. SRD5A3 and SLC4A4 are both recessively inherited genes; however, our patient, and some other patients with mental retardation and/or learning difficulties [8, 9], show heterozygous deletion of both genes. Moreover, some symptomatic patients only harbor the SLC4A4 deletion [10], or no deletion of either gene (DECIPHER 321791), suggesting that a more complicated relationship exists between SRD5A3, SLC4A4 or possibly other unidentified genes in this region, in the process of neurodevelopment.
The common craniofacial features of 4q deletions, which were also observed in our case, are microcephaly and ocular defects such as exophthalmos, astigmatism, strabismus, and cataracts. Microcephaly is known to be associated with CEP135 (MIM*611423), and was identified in patients with autosomal recessive primary microcephaly (MIM#614673) in homozygote form [11, 12]. The microcephaly of our case, being a heterozygous CIP135 deletion, and some other patients, also with the same deletion, may therefore not be explained by CEP135 alone. Haploinsufficiency for BMP3 (MIM*112263) and BMP2K (MIM*617648) is the prevailing hypothesis concerning the origin of ocular anomalies [10]. However, our case did not have a deletion of either of these genes. To date, none of the genes within the deleted region associated with ocular expression have been associated with ocular abnormalities.
Dental and enamel defects, which are also common features of 4q deletions, could be ascribed to haploinsufficiency of the ENAM (MIM*606585), AMBN (MIM*601259) and AMTN (MIM*610912) genes, at 4q13.3. Dental enamel, a highly mineralized tissue, is rigorously controlled in ameloblasts by the interaction of several organic matrix molecules, including enamelin, encoded by ENAM, and ameloblastin, encoded by AMBN. Mutations of the ENAM and AMBN genes cause amelogenesis imperfecta type 1B (MIM#104500), 1C (MIM#204650) and 1F (MIM#616270) [13, 14]. Amelotin, an ameloblast-specific protein, encoded by AMTN and specifically expressed in maturation-stage ameloblasts, and partial deletion of the AMTN gene, was reported in amelogenesis imperfecta type 3B (MIM#617607) [15]. Our case and that reported by Chen et al. [10], both lacking variants in these genes, showed dental disorders, whereas the case reported by Hemati et al. [8] did not show any dental disorders (Table 1). These conflicting findings suggest the possibility of either incomplete penetrance, or that other genes, in the deleted region or elsewhere, likely also play strong roles in amelogenesis.
Haploinsufficiency of KIT (MIM*164920) is well known to be related to piebaldism [10]. Our patient, along with some previously reported patients with proximal 4q deletion, showed KIT deletion without piebaldism [8] (Fig. 3, Table 1). This suggests incomplete penetrance of KIT haploinsufficiency, or the presence of other factors that could affect the expression of piebaldism.
In summary, our review of the present case, and the few previous cases, confirmed with aCGH, indicates that the genes currently considered to be associated with specific phenotypes of 4q12q21.1 deletion cannot sufficiently explain the phenotypes observed or the variations in clinical manifestations among cases, such as mental retardation, microcephaly, ocular anomalies, dental anomaly, and piebaldism. This is in contrast with the conclusions of some previous publications [8-10] and highlights the need for ongoing investigations into the genes associated with 4q12q21.1 deletion to assist in identifying genotype-phenotype associations more clearly.
Go to :
 XML Download
XML Download