

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Autosomal dominant chronic mucocutaneous candidiasis (AD-CMC) is a primary immunodeficiency disorder (PID) characterized by a chronic and recurrent mucocutaneous fungal infection [1]. Genetic mutation of the signal transducer and activator of transcription 1 (STAT1) protein has been known to cause AD-CMC [2]. The STAT1 protein is a member of the STAT family regulated by the Janus kinase (JAK), which is translocated to the nucleus and regulates the expression of genes related to STAT signaling and immune system regulation [3]. In particular, gain-of-function (GOF) STAT1 mutation disrupts interferon-γ, interleukin-17, and interleukin-22 signaling, causing defective Th1 and Th17 responses (Fig. 1A) [2, 4].
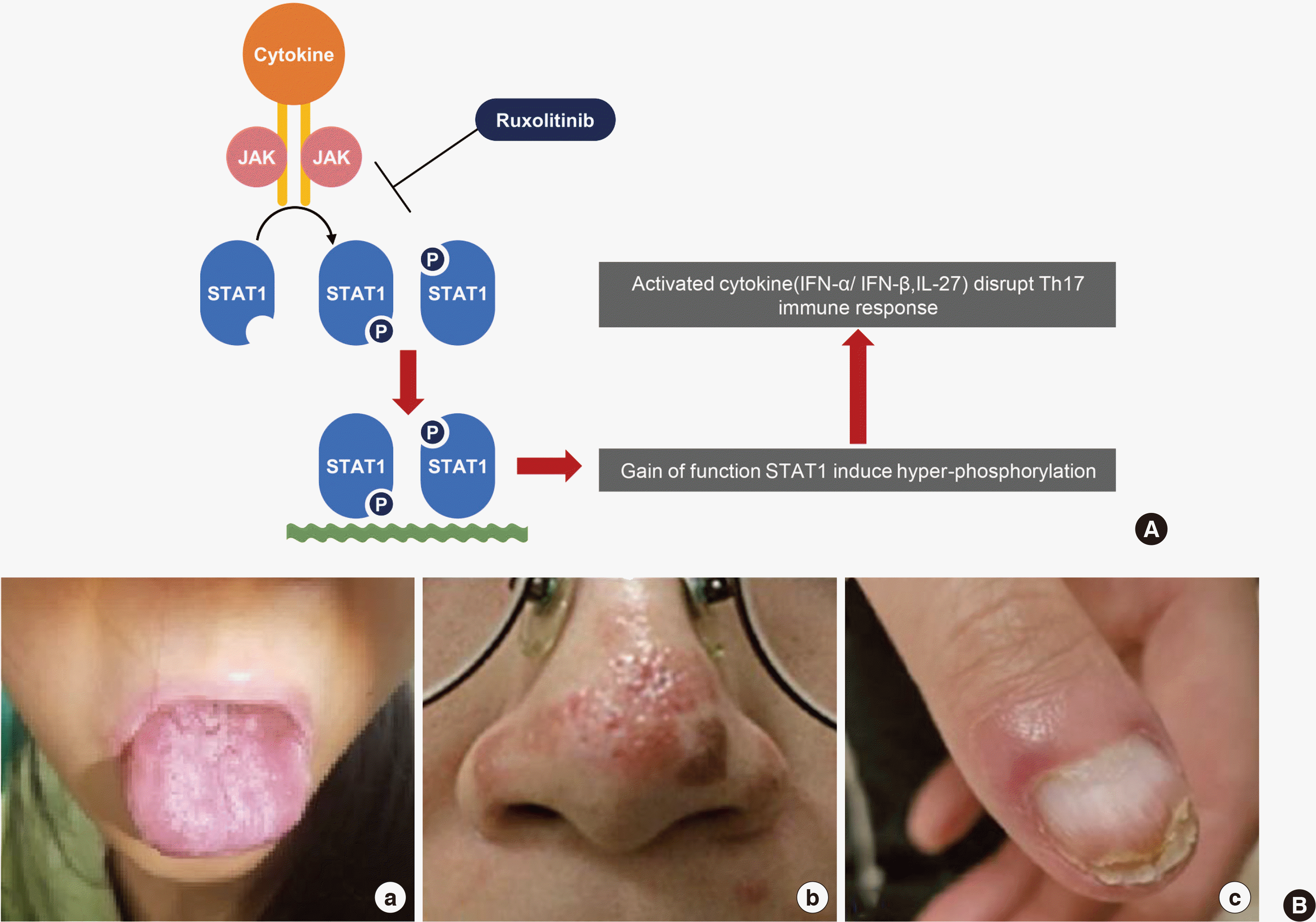
Although GOF STAT1 mutation can be identified by genetic testing or western blotting, both methods are laborious, time-consuming, and expensive [5]. Recently, flow cytometry (FCM) has been increasingly used to measure intracellular phosphorylated-protein levels. Some studies have suggested that FCM could be used as a diagnostic tool to monitor the phosphorylation of STAT1 (pSTAT1) in addition to genetic testing and western blotting [5, 6]. Here, we report a case of GOF STAT1 mutation in a patient diagnosed with AD-CMC by whole-exome sequencing and present the results of FCM, which are concordant with the sequencing results.
Go to : 
CASE REPORT
This case report was approved by the Institutional Review Board of Samsung Medical Center (IRB No. 2018-07-140). In May 2017, a 24-year-old female patient who suffered from M. massiliense infection with onychomycosis and mucocutaneous candidiasis on her nose and mouth for approximately 10 months at another hospital was transferred to the Samsung Medical Center (Seoul, Korea) (Fig. 1B). She experienced recurrent episodes of oral thrush, sinusitis, and pneumonia from early childhood and was diagnosed with bronchiectasis at the age of 12 and pneumonia caused by Pseudomonas aeruginosa at the age of 18. Based on the presumptive diagnosis of PID, immunological work-up such as the lymphocyte subset analysis, IgG subclass, and autoimmune antibody testing with genetic testing of genes such as the cystic fibrosis transmembrane conductance regulator (CFTR) had been performed. Except for the increased erythrocyte sedimentation rate (64 mm/hr), the patient had normal complete blood count. There were no pathogenic variants of the CFTR gene, and the levels of serum IgG (1,580 mg/dL), IgA (90 mg/dL), IgM (94 mg/dL), and alpha-1-antitrypsin (173.7 mg/dL) were normal. Although the level of the rheumatoid factor (77.9 IU/mL) was increased, the results of the fluorescent antinuclear antibody, antineutrophil cytoplasmic antibody, and extractable nuclear antigen testing were negative. The results of the extractable nuclear antigen testing for SS-A, SS-B, ribonucleoprotein, and Smith were also negative. However, a GOF mutation in the STAT1 protein (NM_007315.3:c.800C>T, p.Ala267Val) was detected by a targeted exome sequencing [7].
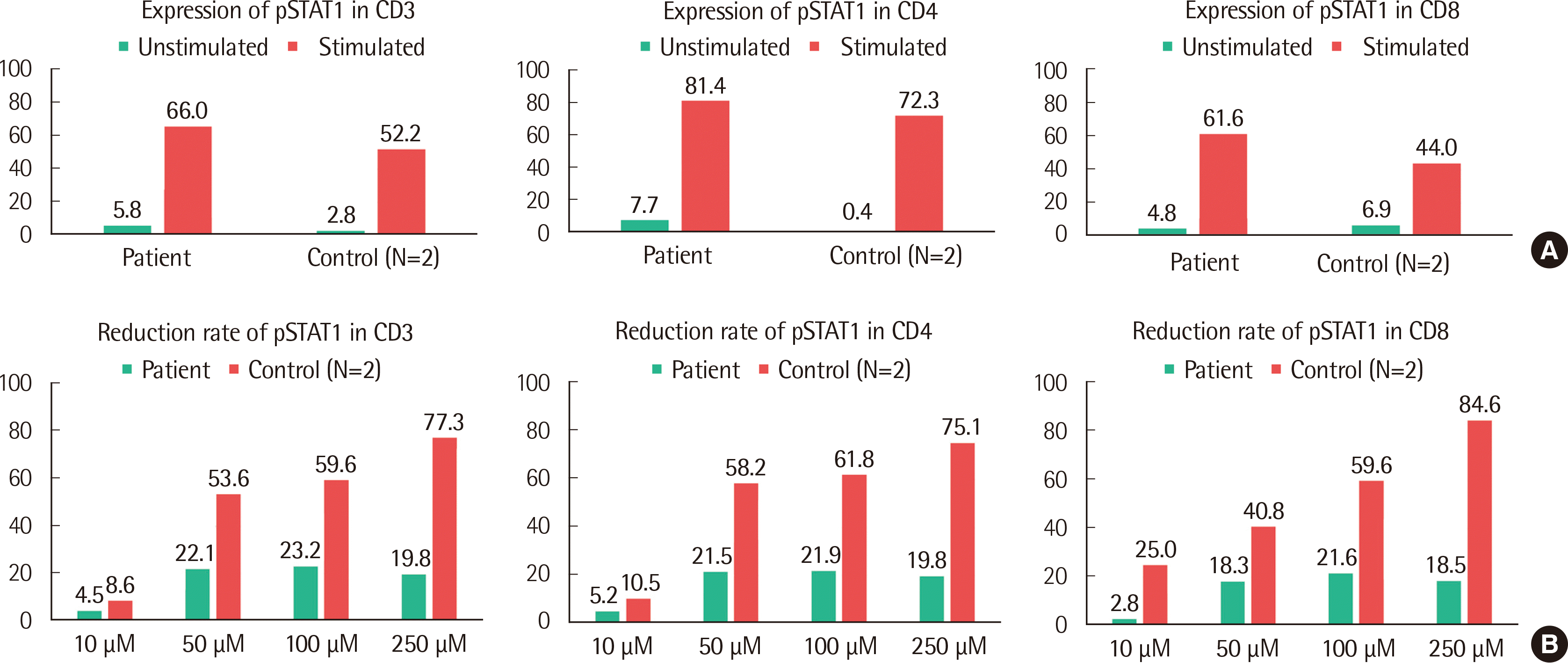
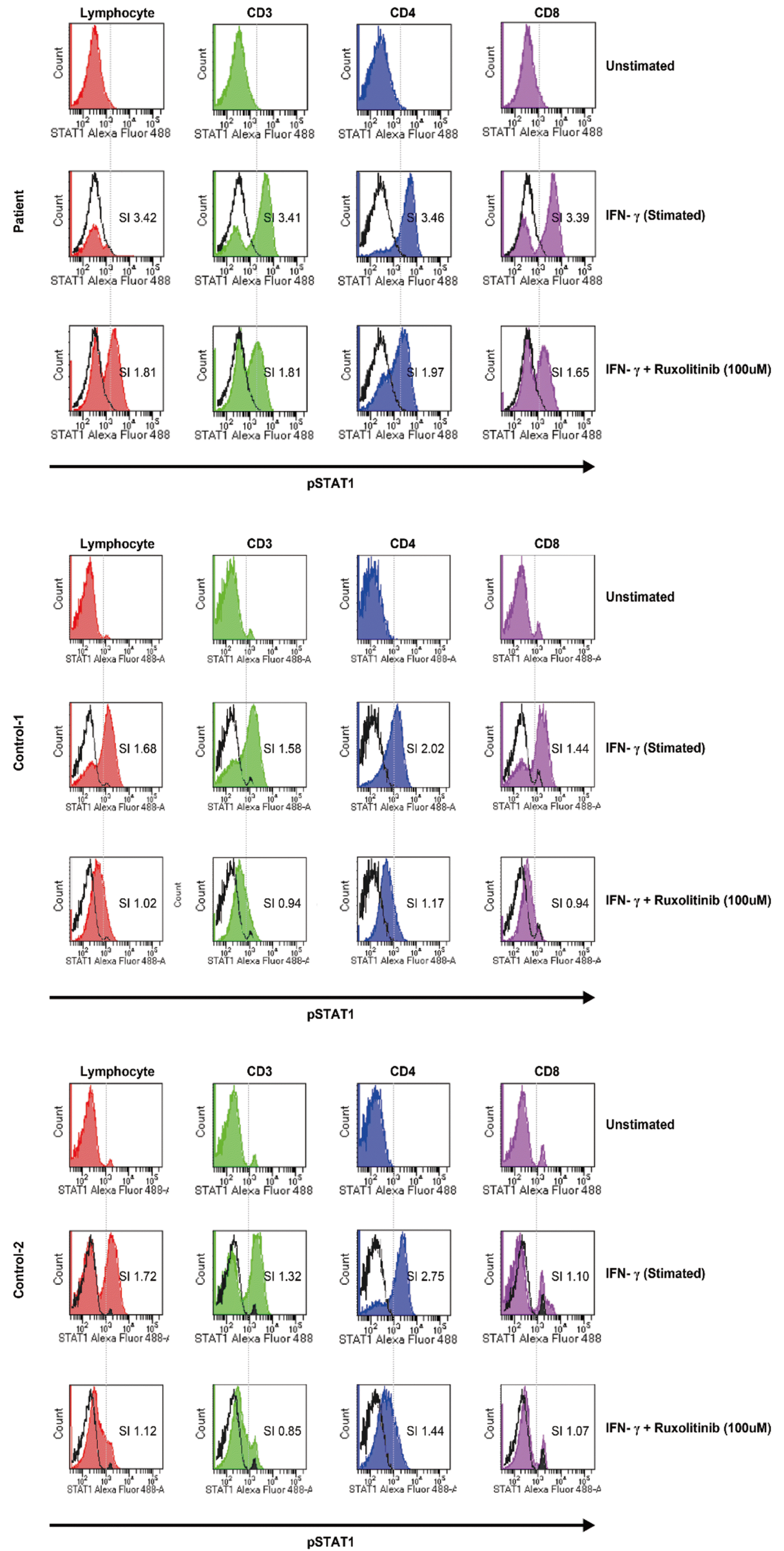
Under the patient’s consent, we investigated the functional consequences of the GOF STAT1 mutation by performing an FCM analysis of the intracellular STAT1 to evaluate whether FCM could be used as a diagnostic and monitoring tool of the STAT1 GOF mutation. Heparin-anticoagulated peripheral blood samples were collected from the patient and normal controls (N=2). Peripheral blood mononuclear cells were acquired by a density gradient centrifugation using Ficoll (General Electric Healthcare Bio-Sciences, Uppsala, Sweden) and stimulated without or with 20 ng/mL interferon-γ (IFN-γ). Ruxolitinib (Novartis Pharmaceutical Corporation, East Hanover, NJ, USA), an inhibitor of JAK1/JAK2, was also added at different concentrations (10 μM, 50 μM, 100 μM, and 250 μM) to observe the inhibition of phosphorylation. The cells were stained with pre-titered Alexa Fluor 488-conjugated pSTAT1 antibody. The stimulation index (SI) was calculated for the gated lymphocytes by dividing the mean fluorescent density (MFI) of stimulated cells by that of the unstimulated cells. Four-color FCM was performed on a FACSCanto Ⅱ apparatus with FACS DIVA (version 8.0) software (all from BD Biosciences, San Jose, USA). When the SI was greater than 2.0, pSTAT1 expression was regarded as significant.
The patient showed a significantly increased MFI, which indicated the expression of pSTAT1 with resistance to the inhibitor compared with the control group. When the CD3+ cells of the patient were stimulated by 20 ng/mL IFN-γ, the SI of pSTAT1 was 3.41 while the mean SI of pSTAT1 was 1.4 in the control group (N=2) (Fig. 2A and Fig. 3). Furthermore, the SI of pSTAT1 was 1.81 when the CD3+ cells of the patient were treated by 100 μM ruxolitinib while the mean SI of the controls was 0.9. The reduction rate of the mean pSTAT1 in the control CD3+ cells was 59.6% and only 23.2% when the patient was treated with 100 μM ruxolitinib (Fig. 2B and Fig. 3, respectively). The results of FCM, which measured the response to IFN-γ without or with ruxolitinib, was in agreement with the pathogenesis of the GOF STAT1 mutation [8]. Although the effect of the GOF STAT1 mutation is not fully understood [1, 9], some studies have recommended ruxolitinib, which inhibits JAK1/JAK2, as a treatment alternative for STAT1 GOF [10-12]. Based on the results of genetic testing and FCM, the patient was treated with ruxolitinib (10 mg, bid) from December 2018 to April 2019. Before she started the medication, the patient had frequent events of abdominal discomfort, oral thrush, and fungal infection even though she was administered 100 mg fluconazole three days a week. After starting ruxolitinib, the patient’s general condition was improved within a week and her oral thrush, along with onychomycosis of a fingernail, was improved after two weeks. During the three-month treatment period, the patient’s general condition including abdominal discomfort was improved. In addition, the patient showed no event of oral thrush, sinusitis, and fungal infection such as candidiasis on her nose or fingernails.
Go to :
DISCUSSION
PIDs are inherited disorders of the immune system that have heterogeneous clinical phenotypes [13]. Because of the phenotypic diversity and rare prevalence rate [14], PID diagnosis is challenging and genetic testing is an essential and a promising diagnostic tool for PIDs [15]. Although genetic testing is crucial to diagnose PIDs, fast and anticipative diagnosis of PIDs is also important to manage and reduce complications associated with the infection. In this respect, FCM has recently been suggested as a diagnostic tool for PIDs [5, 6, 16]. STAT1 is a key transcription factor that mediates the IFN-α/β signaling and regulates the immune response via factors such as IFN-γ and interleukin-17 [3], and both loss- and gain-of-function STAT1 mutations have been described in PIDs [9]. In the case of the GOF STAT1 mutation, STAT1 hyper-phosphorylation causes a defect in Th17 cell differentiation, which is crucial for a mucosal antifungal immunity, and induces clinical characteristics of CMC such as an oral thrush and candida infection of the skin and mucous membranes [17]. Thus, the phosphorylation of STAT1 may be observed with FCM and can be used as a trace marker for the treatment response.
In conclusion, we measured the level of pSTAT1 in a patient who was diagnosed with AD-CMC by genetic testing, and the results of FCM analysis were in agreement with the known pathogenesis of the GOF STAT1 mutation. Compared with the normal control group, the patient showed a significant expression of pSTAT1 (greater than two-fold SI) and a decreased reduction rate in response to the inhibitor. Although it is difficult to determine the cut-off value for MFI or SI for the diagnosis of AD-CMC because of the limited number of pSTAT1 measurements performed using FCM worldwide, we suggest that FCM may be used as a simple and fast supplementary diagnostic tool for the GOF STAT1 mutations [5]. In addition to the potential of FCM as a diagnostic tool, FCM-based phosphorylation assays may be useful for monitoring treatment response in patients harboring a GOF or loss-of-function STAT1 mutations [5, 18]. To the best of our knowledge, this is the first report of pSTAT1 measurement using FCM in Korea.
Go to :
 XML Download
XML Download