

 PDF
PDF Citation
Citation Print
Print



서 론
완간역위(pericentric inversion)는 양 절단점 사이에 염색체 중심절을 포함한 절단부가 회전하여 장완과 단완의 일부가 재배열된 6338염색체의 구조적 이상이다. 대부분의 완간역위는 균형재배열로 보인자의 표현형이 정상이다. 하지만, 생식세포의 감수분열 과정 중에 역위 고리(inversion loop)를 형성해 염색체의 결실 혹은 중복을 보이는 불균형재배열 재조합염색체가 형성될 수 있다. 완간역위를 가지고 있는 여성의 경우 불균형재배열 재조합염색체로 인해 비정상 표현형을 지닌 신생아가 출생할 위험도가 7%이고, 남성의 경우 5%의 위험도를 지니고 있다[1].
Beckwith-Wiedemann 증후군(BWS, OMIM #130650)은 가장 흔하게 발생하는 과성장 증후군(overgrowth syndrome)이고 배양성종양(embryonal tumor)의 높은 발생 가능성을 특징으로 한다. BWS는 11p15.5 부위에 위치한 IGF2와 CDKN1C와 같은 각인된 성장 조절 유전자(imprinted growth regulatory genes)의 발현 양상의 변화 때문에 발생한다. 이러한 변화의 원인과 빈도는 각각 imprinting center 2 (IC2)의 메틸화 소실이 50%, 부계의 uniparental disomy (UPD)가 20%, imprinting center 1 (IC1)의 과메틸화가 5%, CDKN1C의 병원성 변이가 5%이다[2-4]. Jacobsen 증후군(JBS, OMIM #147791)은 11qter의 결실 때문에 발생하는 인접 유전자 증후군(contiguous gene syndrome)으로 특징적인 이형 양상(dysmorphic features), 저성장, 발달장애, 혈소판 감소증과 심장, 신장, 위장관 등의 기형(malformation)과 같은 다양한 임상양상을 보인다. 절단점은 주로 11q23.3에서 생겨 결실이 말단부까지 이어지는데 결실의 크기는 7 Mb에서 20 Mb로 다양하고 결실의 크기가 클수록 질병의 임상 양상도 심해진다[5, 6]. 저자들은 아버지의 inv(11)(p15.1q24.2)로 기인한 11pter 삼염색체(trisomy)와 11qter 단일염색체(monosomy)로 인해 Beckwith-Wiedemann 증후군과 Jacobsen 증후군이 진단된 환자를 보고하고자 한다.
Go to : 
증 례
환자는 재태연령 35주 2일에 양수과다증으로 제왕절개술로 태어난 여아로 출생 직후 몸무게 2,940 g (90th percentile), 신장 48 cm (50th percentile), 두위 34 cm (90th percentile)이었다. 환아는 산전 초음파상 이중출구우심실(double outlet right ventricle, DORV), 심실중격결손(ventricular septal defect, VSD), 폐형성저하증(lung hypoplasia), 전신의 피부부종, 양수과다증, 내반족(clubfoot) 소견이 관찰되었다. 출생 직후 마스크 양압환기법 적용에도 호전되지 않는 청색증 및 호흡곤란으로 중환자실에 입실하였다. 영상 검사에서 심실중격결손을 동반한 팔로형 이중출구우심실(Fallot DORV with VSD), 중심폐동맥폐쇄(interruption of central pulmonary artery), 양측 동맥관개존증(bilateral patent ductus arteriosus), 다낭신장병(polycystic kidney disease), 뇌량 저형성(hypoplastic corpus callosum)이 관찰되었고 뇌전도검사에서 정상소견을 보였다. 신체 검진에서 안검열의 외하방 경사(down-slanting palpebral ¬ssure), 물갈퀴목(webbed neck), 처진귀(low set ears), 넓은 유두 간격, 양측 5번째 발가락의 측만지증(clinodactyly of both 5th toe)이 있었다. 환아는 출생 직후 시행한 complete blood count (CBC)에서 WBC: 3,400/μL, hemoglobin: 14.9 g/dL, platelet: 73,000/L를 보였고, 12개월 시행한 CBC는 WBC: 2,100/μL, hemoglobin: 10.7 g/dL, platelet: 83,000/μL, 16개월에는 WBC: 2,900/μL, hemoglobin: 10.6 g/dL, platelet: 59,000/μL를 보였다. 신생아 중환자실에 입실 후 1개월과 4개월째에 중심정맥카테터 관련 균혈증으로 항생제 치료를 받았다. 생후 2개월에 이중출구우심실의 전체 복원, 폐동맥 재건술, 동맥관개존증 수술을 받았다. 생후 3개월에 시행한 복부 초음파 검사에서 9.2 cm의 비장비대(normal range at 3-6 month; (female) 4.5-5.6 cm/(male) 4.9-7.0 cm)가 보였고 TSH 상승 소견으로 선천성 갑상선저하증이 의심되었다. 생후 4개월에 시행한 청력검사에서 양측이 90 dB의 고도 난청을 보여 청력이 소실되어 보청기를 착용하였다. 경관급식(G-tube feeding)에서 경구급식(oral feeding)으로 변경하면서 구토 증세가 발생하여 촬영한 복부 초음파 검사에서 비대유문협착증(hypertrophic pyloric stenosis)이 확인되어 유문근층절개(pyloromyotomy)를 시행했다. 생후 12개월에 시행한 베일리 영유아 발달검사에서 인지지수(mental development index) 50점 미만, 동작지수(psychomotor development index) 50점 미만으로 원점수에 따른 발달연령은 대근육운동 1-2개월, 소근육운동 3개월, 인지 1개월, 사회성 1-2개월 수준으로 모든 영역에서 발달 지연이 관찰되었다. T-cannula 삽입과 양측 청력 소실로 언어 영역의 평가는 이뤄지지 않았다. 생후 16개월의 신체계측에서 몸무게 9.8 kg (50th percentile), 신장 76 cm (25th percentile)이었다. 본 증례의 임상 양상을 이전에 보고된 다른 증례와 더불어 BWS와 JBS의 대표적인 임상 양상과 비교하여 Table 1에 정리하였다[3, 5, 13].
Table 1
Comparison of clinical features in Beckwith-Wiedemann syndrome and Jacobsen syndrome and Beckwith-Wiedemann and Jacobsen syndromes in a previously reported case and in this case
| Clinical features | Beckwith-Wiedemann syndrome [3] | Jacobsen syndrome [5] | Previously reported case [13] | Our patient |
|---|---|---|---|---|
| Prenatal abnormalities | Macrosomia | Intrauterine growth retardation | Macrosomia | Macrosomia |
| Polyhydramnios | Premature birth | Polyhydramnios Premature birth | ||
| Premature birth | Polyhydramnios | |||
| Growth | Generalized overgrowth (macrosomia) | Height; <10th percentile in 75% | Height; +1.5 SD | Height; 25th percentile |
| Weight; <10th percentile in 58% | Weight; +3.5 SD (at birth) | Weight; 50th percentile (at 16 months) | ||
| Developmental abnormalities | Developmental delay | Psychomotor retardation | Psychomotor retardation | Psychomotor retardation |
| Dysmorphic features | Macroglossia | Down-slanting palpebral fissure | Down-slanting palpebral fissure | Down-slanting palpebral fissure |
| Ear abnormalities | Flat nasal bridge | Ocular hypertelorism | Webbed neck | |
| Cleft palate | Short neck | Flat nasal bridge | Low set ears | |
| Hemihyperplasia | Low set ears | Short neck | Fifth-finger clinodactyly | |
| Webbed neck | Low set ears | Clubfoot | ||
| Fifth-finger clinodactyly | Fifth-toe clinodactyly | |||
| Clubfoot | ||||
| Neurological abnormalities | Hearing loss | Hearing loss | Hypotonia | Hearing loss |
| Posterior fossa abnormalities | Agenesis of the corpus callosum | Hypoplastic corpus callosum | ||
| Cardiac abnormalities | Cardiomegaly | Ventricular septal defect | - | Double-outlet right ventricle, Ventricular septal defect |
| Hypoplastic left heart syndrome | ||||
| Abdominal abnormalities | Hepatomegaly | Pyloric stenosis | Pyloric stenosis | Pyloric stenosis |
| Omphalocele | Feeding difficulties | Omphalocele | Splenomegaly | |
| Umbilical hernia | ||||
| Diastasis recti | ||||
| Renal abnormalities | Nephromegaly | Unilateral kidney dysplsia | - | Polycystic kidney |
| Nephrocalcinosis | Polycystic kidney | |||
| Medullary dysplasia | ||||
| Neoplasia | Wilms tumor | - | - | ND |
| Nephroblastomatosis | ||||
| Neuroblastoma | ||||
| Rhabdomyosarcoma | ||||
| Hepatoblastoma | ||||
| Adrenal carcinoma | ||||
| Laboratory finding | Hyperinsulinism | Thrombocytopenia | Thrombocytopenia | Thrombocytopenia |
| Transient hypoglycemia | Anemia | Anemia | Anemia | |
| Hypothyroidism | Neutropenia | Neutropenia | Neutropenia | |
| Polycythemia | Recurrent infections | Recurrent infections | Recurrent infections | |
| Hypothyroidism | ||||
| Molecular etiology | Molecular alterations in growth regulatory genes on chromosome 11p15.5 | Terminal deletion of the long arm of chromosome 11 | Duplication at 11p15.5p15.3 | Duplication at 11p15.5p14.3 |
| Deletion at 11q24.1q25 deletion | Deletion at 11q23.3q25 |
![]()
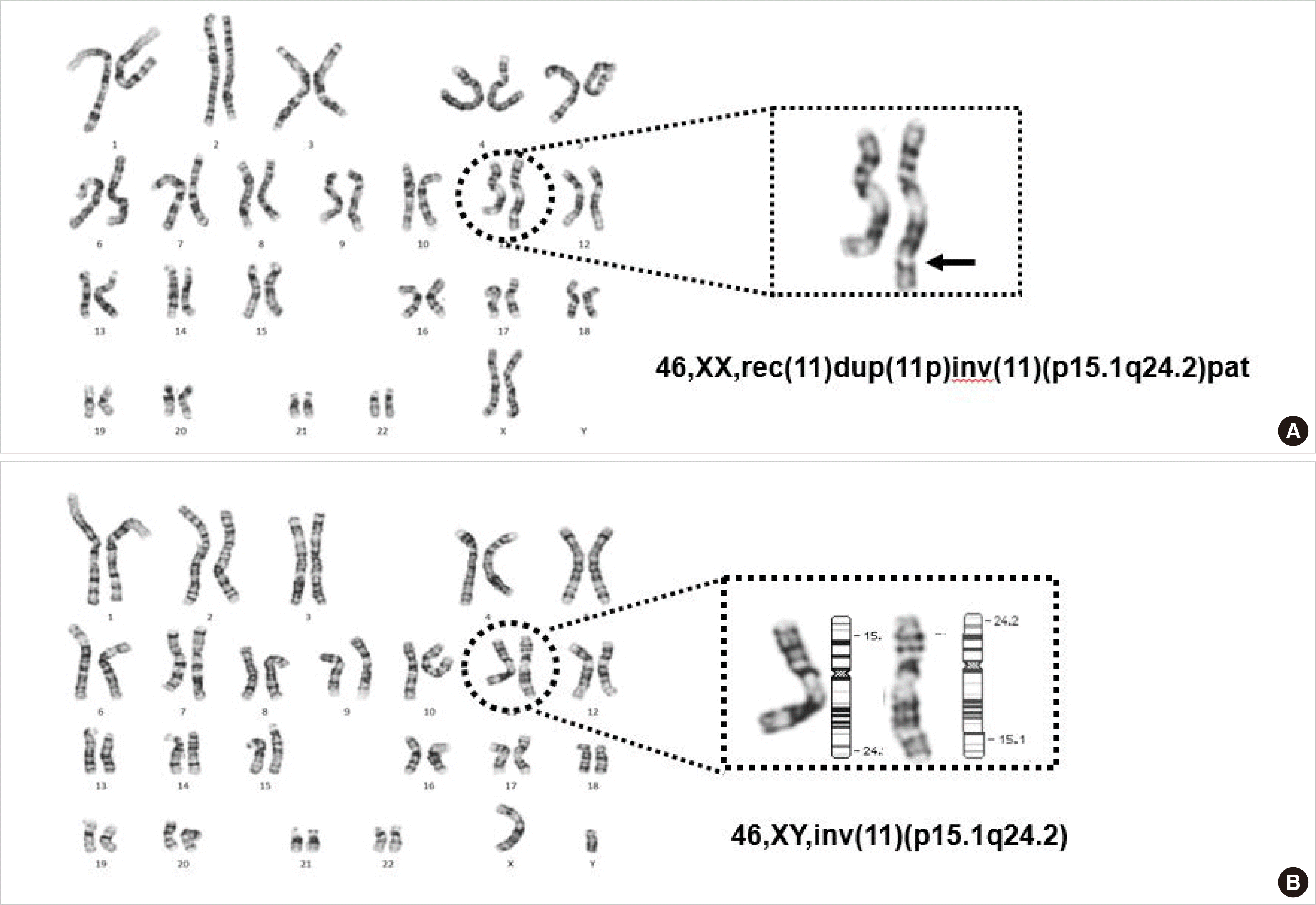
출생 직후 말초혈액으로 시행한 염색체검사에서 46,XX,der(11) t(11;?)(q23.3;?)로 11q의 불균형전좌가 보였다(Fig. 1A). 부모의 염색체검사에서 어머니의 핵형은 정상이었으나 아버지의 핵형이 46,XY, inv(11)(p15.1q24.2)로 완간역위가 있는 염색체의 균형재배열이 관찰되었다(Fig. 1B). 따라서 환아 핵형의 der(11)은 아버지에서 유래한 inv(11)(p15.1q24.2)의 감수분열 시 재조합에 의해 발생한 rec(11) dup(11p)inv(11)(p15.1q24.2)로서 11번 단완(p15.1-pter) 중복 및 11번 장완(q24.2-qter)의 결실을 보였다.
SALSA MLPA (multiplex ligation-dependent probe ampli¬cation) ME030 BWS/RSS probemix (MRC-Holland, Amsterdam, Netherlands)를 이용한 methylation-speci¬c (MS)-MLPA 검사를 통해 11p15.5 부위에 위치한 H19와 KCNQ1OT1의 결실 및 중복과 메틸화 양상을 검사했다. 검사 결과에서 H19와 KCNQ1OT1가 중복되었고, IC1의 과메틸화 소견과 IC2의 정상 메틸화 소견을 보여, 부계의 11p15.5 부위가 중복된 것임을 확인할 수 있었다(Fig. 2).
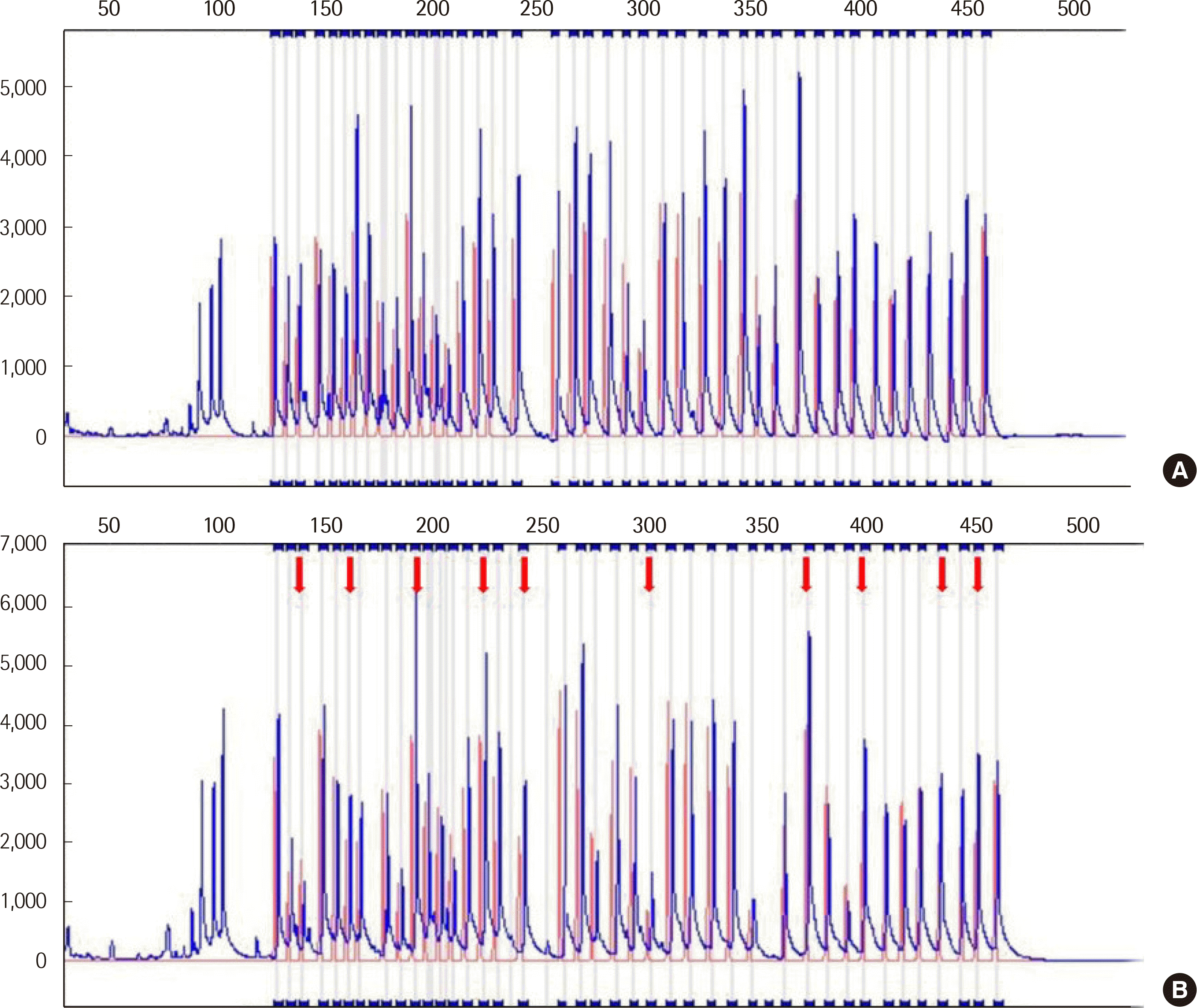 | Fig. 2Capillary electrophoresis electropherogram of MS-MLPA for 11p15.5 region. The red peak represents control DNA amplification. The blue peak represents the patient’s DNA amplification. (A) Detection of copy number change without HhaI enzyme. Peak heights of all probes targeting H19 and KCNQ1OT1 increased, showing duplication of H19 and KCNQ1OT1. (B) Detection of methylation status with HhaI enzyme. The red arrows indicate increased peak heights of probes targeting H19, showing hypermethylation of imprinting center 1.
Abbreviation: MS-MLPA, methylation-specific multiplex ligation-dependent probe amplification.
|
Cytoscan 750K Array (Affymetrix, Santa Clara, CA, USA)를 이용한 염색체 마이크로어레이검사에서 GRCh37 (Genome Reference Consortium Human Build 37)을 기준으로 11p15.5p14.3(23068_ 23885320)에서 23.8 Mb 크기의 중복과 11q23.3q25(121090596_ 134937416)에서 13.8 Mb 크기의 결실을 확인하여(Fig. 3, Table 2), 최종적으로 ISCN 2016에 따라 46,XX,rec(11)dup(11p)inv(11)(p14. 3q23.3)pat.arr[GRCh37] 11p15.5p14.3(23068_23885320)X3,11q23. 3q25(121090596_134937416)x1로 기술하였다.
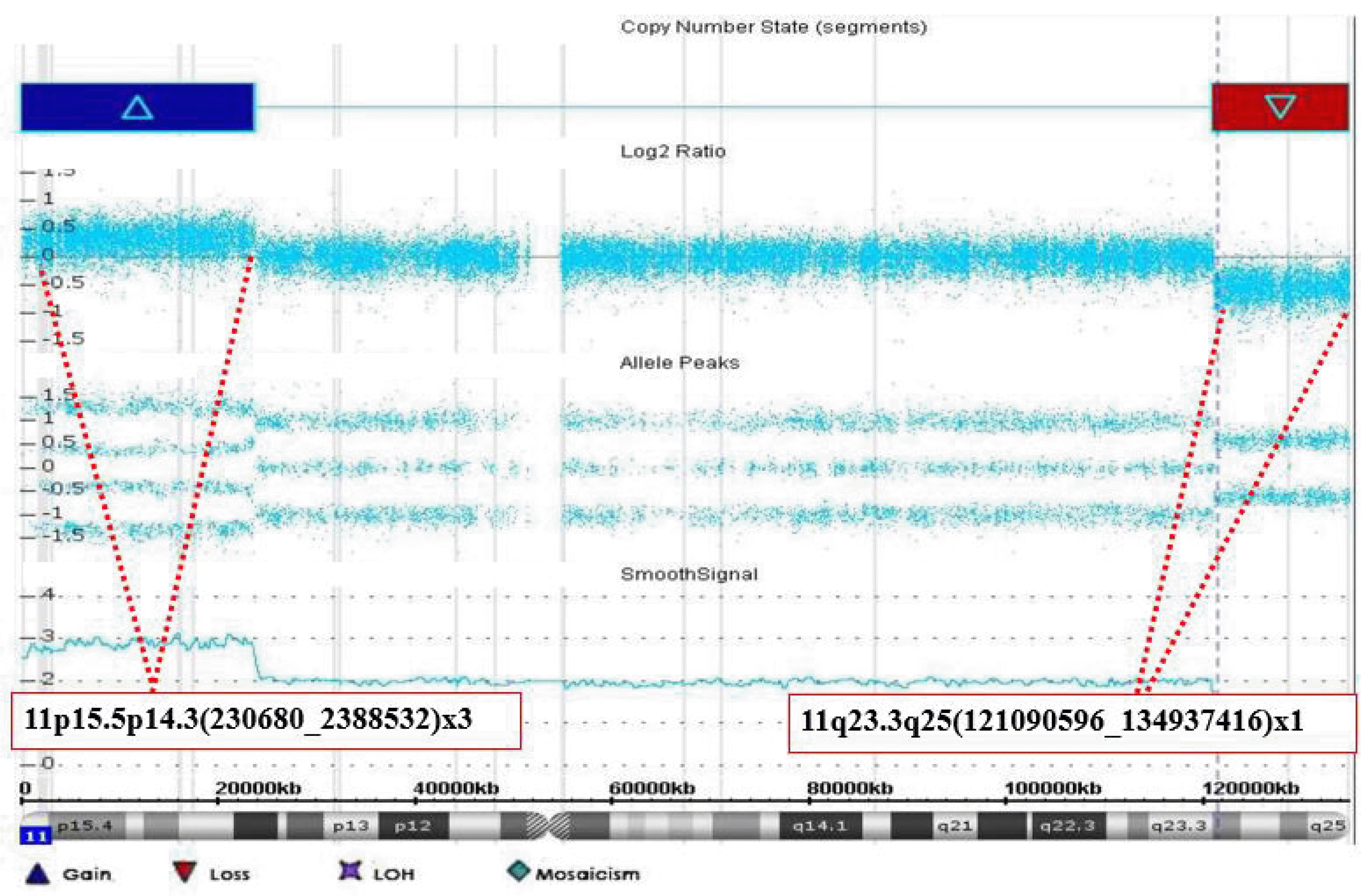 | Fig. 3Duplication and deletion at 11p and11q in this patient demonstrated by chromosomal microarray. The blue bar and red bar indicate duplication and deletion regions, respectively. The chromosomal microarray of chromosome 11 showed a 23.8 Mb duplication at 11p15.5p14.3 and a 13.8 Mb deletion at 11q23.3q25.
|
Table 2
Location, size, and syndrome-related OMIM genes of the copy number variants detected in the chromosomal microarray
![]()
Go to :
고 찰
본 증례는 산전 초음파 검사에서 확인된 다양한 기형을 가진 환아의 출생 후 염색체검사에서 11번 염색체의 비정상 소견이 관찰되어 시행한 환아의 부모 염색체검사 결과, 환아의 비정상 11번 염색체는 11번 염색체의 완간역위가 있는 아버지로부터 유래하여 11pter 중복과 11qter 결실을 보이는 재조합염색체로 확인되었음을 보여주고 있다. 추가적으로 시행한 MS-MLPA와 염색체 마이크로어레이검사에서 부계의 11p15.5p14.3 중복과 IC1의 과메틸화 양상을 검출함으로써 11p15.5의 부계 UPD가 발병 원인 중의 하나인 Beckwith-Wiedemann 증후군으로 진단할 수 있었다. 또한 11q23. 3q25의 13.8 Mb 결실도 확인하였으며, Beckwith-Wiedemann 증후군과 Jacobsen 증후군 임상양상을 보이는 본 환아에게서 두 질환이 인접 유전자 증후군(contiguous gene syndrome)으로 발현되었음을 확인할 수 있었다.
BWS의 발병 기전은 크게 4가지로 모계 염색체 IC1의 과메틸화, 모계 염색체 IC2의 메틸화 소실, 부계의 UPD, 그리고 CDKN1C의 병원성 변이로 구별할 수 있다[3]. 본 환아의 경우 아버지에서 유래한 재조합염색체에 중복된 11p15.5 부위가 있어 부계의 UPD와 같은 발병 기전을 지닌다. 중복이 발생한 부위에 존재하는 OMIM에 등재된 유전자 중에서 HRAS, IGF2, PNPLA2, H19, KCNQ1OT1가 BWS의 임상 양상과 관련이 있다(Table 2). 특히, IGF2는 11p15.5에 위치해 각인된 유전자에 의해 발현이 조절되는 유전자로 BWS와 관련된 중요한 유전자이다[7, 8]. 환아에서 관찰된 양수과다, 비장종대, 갑상선저하증, 발달지연 등이 BWS에서 보이는 임상 소견이었다. 환아에서 BWS의 특징적인 소견인 거체구증(macrosomia)은 뚜렷하지 않았지만 이는 환아가 동시에 가진 JBS의 성장 지연 때문으로 보인다.
본 환아는 11pter과 더불어 11qter의 결실이 있는데 이는 JBS와 관련이 있다. 결실의 절단점은 11q23.3로서 결실된 크기는 13.8 Mb이었다. 결실이 발생한 부위에 존재하는 OMIM에 등재된 유전자 중에서 FLI1, JAM3, KCNJ1, KCNJ5가 JBS의 임상 양상과 관련이 있다(Table 2). 대부분의 JBS 환자는 거대핵구형성이상(dysmegakaryopoiesis)과 더불어 혈소판 감소증을 보인다. 또한, 비정상적인 혈소판내 알파입자(alpha granules) 간 융합으로 입자 내 응고와 관련된 성분들의 분비가 감소해 혈소판의 기능도 같이 감소해 있다[5, 6]. FLI1은 거대핵구의 형성과 분화에 중요한 역할을 하며 FLI1 유전자결손 동물모델의 골수에는 거대핵구가 없거나 감소되어 있고 소형거대핵구의 소견을 보인다는 연구 결과가 있고, JAM3, ETS1, NFRKB 유전자들도 혈소판 기능에도 관여한다고 알려져 있다[6, 9, 10]. JBS 환자에게 지적 장애는 흔히 관찰되는 임상 양상 중 하나이다. 지적 장애의 수준은 결실 크기에 관련이 있어 크기가 클수록 증상이 심해진다[6]. 또한 심장 기형도 JBS 환자의 56%의 경우에서 관찰된다. 심장 기형과 관련된 유전자들로 SNX19, ADAMTS8, JAM3, ROBO4, KCNJ5 등이 알려져 있다[10, 11]. 유문협착증은 소화기 기형 중 56%의 빈도로 가장 흔하게 관찰되는데[6] 본 환아도 생후 4개월에 비대유문협착증으로 유문근층절개를 받았다. 신장 기형도 JBS 환자의 8%에서 관찰되는데 신장 형성이상(kidney dysplasia), 다낭신장(polycystic kidney) 등이 있다. 신장 기형과 관련된 유전자로 KCNJ1과 ADAMTS15가 알려져 있다[5, 6].
11p15.5의 중복과 11qter의 결실로 인해 BWS와 JBS가 동시에 진단된 증례는 현재까지 국내에는 보고된 적은 없다. Gadzicki 등은 본 환아와 같이 부계의 완간역위로 인해 중복과 결실이 생긴 증례를 보고했다[12]. Putoux 등은 11pter 삼염색체증과 11qter 단일염색체증이 섞임증(mosaicism) 형태로 존재하는 환아의 증례를 보고했다[13]. 두 증례 모두 임상 양상과 더불어 염색체검사와 마이크로어레이 검사를 통해 환자의 진단이 이루어졌다. 부계의 완간역위가 UPD를 초래해 BWS의 원인이 되고 이와 같은 경우 JBS가 동반될 수 있기 때문에, 염색체검사와 더불어 MLPA, 마이크로어레이검사 등 가능한 방법을 시행해 관련 부위의 중복 및 결실 여부를 확인하는 것이 진단에 도움이 된다. 또한 부모가 완간역위의 보인자인 경우 불균형재배열의 임신 위험이 있다. 본 환아의 부모에서는 향후 임신시 재발 위험도와 착상전유전자검사 등에 대한 유전상담이 시행되었다.
Go to :
 XML Download
XML Download