

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Gorlin–Goltz syndrome, also known as the basal cell nevus syndrome, is an autosomal dominant inherited syndrome that predisposes the patient to the formation of basal cell carcinomas, odontogenic keratocysts, and skeletal anomalies. Pathogenic vari-ants of several genes associated with the sonic hedgehog (SHH) signaling pathway, including PTCH1, have been identified in patients with the Gorlin–Goltz syndrome, and are presumed to cause the disease [1]. The genes responsible for this syndrome were found to be PTCH1 and SUFU by homologs of the Drosophila patched gene [2, 3]. A PTCH2 gene, close homolog of PTCH1, is found in vertebrates. Human PTCH2 has 22 coding exons, which encode a protein of 1209 amino acid residues. A Chinese family and a Japanese girl have been previously reported to possess a pathogenic variant of the PTCH2 gene [4, 5]. To the best of our knowledge, we report the first case of the Gorlin–Goltz syndrome associated with a frame-shift pathogenic variant of PTCH2, in Korea.
Go to : 
CASE REPORT
A 30-year-old woman visited our genetic counseling center for a pregnancy consultation as she was worried about having undiagnosed diseases. She had multiple calcifications on her nose, knee, and intermalleolar space for the past 10 years. For 10 years, she had consulted a doctor who eventually recommended her to receive genetic counseling for an appropriate diagnosis. When the patient came to our tertiary center, a simple X-ray confirmed that she had multiple tiny calcified opacities in the anterior subcutaneous layer of her proximal lower legs and an incompletely fused lateral malleolar growth plate, as well as juxtacortical nodular calcified opacities at their lateral aspect (Fig. 1A, B). Moreover, computed tomography (CT) analysis revealed a nodular calcification in the nasal cartilage on the axial plane at the maxillary sinus level (Fig. 1C). We decided to send her peripheral blood sample to the Baylor College of Medicine (Houston, TX, USA) for whole exome sequencing. The patient did not present the pathogenic variants of the PTCH1 or SUFU genes. However, a heterozygous 2-base-pair deletion, c.1172_1173delCT, was detected in exon 9 of the PTCH2 gene; NM_003738.4:c.1172_1173del, p.(Ser391*). This variant was confirmed by Sanger sequencing. This pathogenic variant caused a frame-shift and premature termination at the deletion site, resulting in a truncated form of the protein, PTCH2 p.Ser391*. The patient was eventually diagnosed with the Gorlin–Goltz syndrome, but did not undergo any familial genetic test.
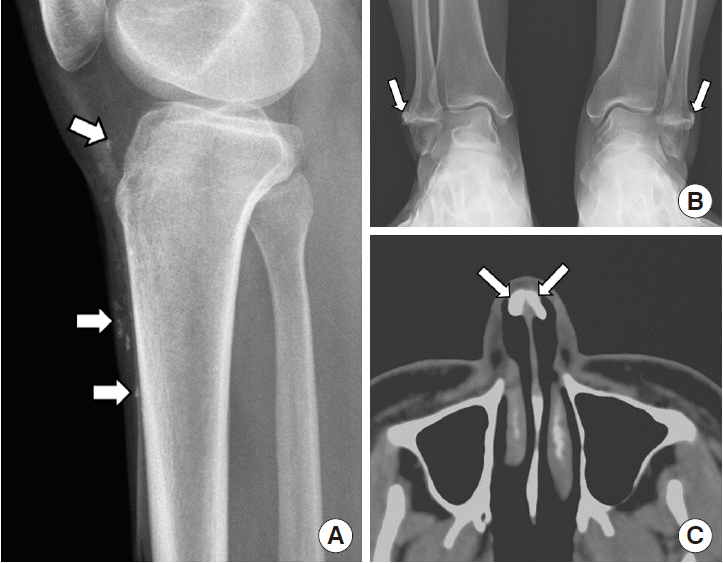 | Fig. 1(A) Lateral view of proximal lower leg plain radiograph shows multiple tiny calcified opacities at anterior subcutaneous layer. (B) Anterior–posterior view of both ankle plain radiographs reveals incomplete fused lateral malleolar growth plates and juxtacortical nodular calcific opacities at their lateral aspect. (C) Computed tomography scan with axial plane of maxillary sinus level indicates a nodular calcification at the nasal cartilage.
|
After diagnosis, she asked us whether this condition would hamper her fertility or pregnancy. We informed her about the disease and suggested appropriate management. We eventually recommended her to visit a doctor specializing in obstetrics and gynecology for further examination and consultation regarding her birth related queries. Fortunately, she neither had ovarian fibromas nor did she need ovary preservation.
Go to :
DISCUSSION
Gorlin–Goltz syndrome is a rare disease that is known to be caused by the pathogenic variants of the PTCH1 and SUFU genes. A missense pathogenic variant of PTCH2 in a Chinese family, and a frame-shift pathogenic variant of PTCH2 in a Japanese girl have been previously reported [4, 5]. In this report, we observed a case with the exact same pathogenic variant as that reported for the Japanese girl [4], which could be a third germ-line pathogenic variant of PTCH2, c.1172_1173del. This pathogenic variant created a premature termination codon at the deletion site in the mutant allele, resulting in the truncation of the PTCH2 protein. The report on the Japanese girl mentioned that she had keratocystic odontogenic tumors and a bifid rib anomaly, which were not present in our patient, who only had multiple calcifications; however, both phenotypes were milder than the typical Gorlin–Goltz syndrome caused by PTCH1 gene variants.
The American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) guideline suggests that variants with an odds ratio more than 4.0 are counted as mutations. We searched the odds ratio for the rs56126236 vari-ant as it was present in the database containing information on healthy population variants. Only two reports for PTCH2 gene variants were found in PUBMED search. One case in Japan was reported to possess this variant in PTCH2 gene [4], and one Chinese family was reported to have different variants in the gene [5]. According to the Genome Aggregation database (gnomAD), the allele frequency of the rs56126236 is single nucleotide polymorphism (SNP) 0.316% (63/19,948) in the East-Asian population. However, the allele frequency is reported as 0.48% (6/1,238) in the Korean reference genome database (KRGD) of Center for Disease Control and Prevention (http://coda.nih.go.kr/coda/KRGDB/index.jsp), which contains genetic information on 622 healthy individuals. The odds ratio is calculated as (a×d)/(b×c), and was found to be 316.6 (confidence interval 19.59–511,852) in case of the East Asian data and 206.3 (confidence interval 11.52–3,696.24) in KRGDB, respectively. The prevalence of this disease is known to be 1 in 57,000 [6]. Considering this rare prevalence and limited cases by pathogenic variants of PTCH2 gene, the calculated odds ratios indicate that this variant could be a causative variant which affects the disease, based on the ACMG/AMP guidelines [7]. Pene-trance of the Gorlin–Goltz syndrome and the related mild phenotypes as observed in this report and in the Japanese report could be considered for this relatively high allele frequency (0.48%).
A genotype–phenotype correlation in the Gorlin–Goltz syndrome remains unclear [8]; however, the pathogenic variant of the SUFU gene is frequently reported in patients with medulloblastoma [9]. Moreover, numerous deletions in PTCH1 manifest atypically, presumably due to the effect on the adjacent genes [10]. A Japanese research group has suggested that the Gorlin–Goltz syndrome in patients with PTCH2 pathogenic variants could be milder phenotype than that in patients with PTCH1 pathogenic variants based on the number of major/minor criteria analyzed by a nationwide survey in Japan [4]. Our patient met only one major criterion, that is, multiple calcifications, and did not present jaw keratocysts or macrocephaly. Therefore, we could presume that the phenotype of the PTCH2 gene pathogenic variant is different from that of the classical phenotype presented by the PTCH1 gene pathogenic variant.
In the present case, the main reason why the patient considered genetic counselling was to determine whether she would face any complications during pregnancy and delivery. In a previous case report, a patient suffering from the Gorlin–Goltz syndrome had a normal pregnancy and labor [11]; however, it is ad-visable to receive appropriate genetic counseling in order to stay informed on the inheritance of genetic disorders.
Go to :
 XML Download
XML Download