

 PDF
PDF Citation
Citation Print
Print


To the editor:
Chromosomal microarray-based copy number variation (CNV) analysis is a first-tier genetic test for patients with intellectual disabilities or autism spectrum disorders and has an overall diagnostic yield of 15-20% [1, 2]. G-banded karyotyping is routinely used for CNV detection, but suffers from low resolution and a detection rate of less than 3% [1]. Recently, next-generation sequencing (NGS) has become an efficient tool for comprehensive characterization of CNVs through generation of numerous short reads in a single run [3]. NGS has increased the throughput of read generation and base-calling accuracy [3] and has recently been employed as a useful diagnostic approach for studying CNVs in various diseases [4-6]. However, using NGS is expensive for routine diagnosis, and the longer turn-around time is not ideal for clinical implementation [7]. Here, we describe a patient with intellectual disabilities, intractable epilepsy, and autistic features for whom low-depth whole-genome sequencing (WGS) has succeeded in detecting pathogenic CNV. This method only requires a small number of reads; thus, it is cost-effective and achievable within a clinical timeframe [7, 8].
The patient was a 14-year-old male with a clinical history of infantile hypotonia. He was born after 38 weeks of gestation and had no known family history of genetic diseases. He had notable facial dysmorphism, with a long face, large ears, and a depressed nasal bridge. The patient exhibited global developmental delays and developed seizures at the age of 48 months, which were treated with valproic acid and were well controlled without any side effects for approximately 8 years. However, generalized tonic-clonic type seizures resumed at 12 years of age. Compared to earlier seizures, the frequency at this age increased and new seizure types such as focal clonic and atonic seizures appeared. These seizures were not well controlled by multiple antiepileptic drugs. In his electroencephalogram, both frontal dominant rhythmic spikes and wave discharges with disorganized background rhythms were frequently seen. Presently, his daily living activities are entirely dependent on others (total dependency level), and he cannot utter any meaningful words. He has a tendency to repeat certain behaviors, such as clapping or swinging his hands and arms. He can walk independently, but an ataxic gait and hamstring tightness have been detected. He has been admitted to the hospital several times for both status epilepticus and severe infections (such as pneumonia), disseminated intravascular coagulation, constipation, and septic shock. His parents and older sister are phenotypically normal and show no evidence of cognitive impairment, hypotonia, or recurrent infections.
To identify causes underlying this disease, we performed genetic testing with approval from the Soonchunhyang University Bucheon Hospital institutional review board (no. 2017-05-032). Cytogenetic analysis revealed a normal karyotype for the patient. After obtaining informed consent, WGS-based CNV analysis was performed at Green Cross Genomic Inc. (Yongin, Korea). Briefly, genomic DNA was extracted from peripheral-blood leukocytes in the patient, sheared to a target size of 250 bp, and sequenced with the NextSeq 500 platform (Illumina, CA, USA) using the 75 bp single-read mode. CNV calling from 3.1 million sequence reads was carried out using DNAcopy software, version 1.38 [9]. Using target-specific reference mapping ratio values of the target sample and normal controls, we calculated the copy number of the target sample. The February 2009 human reference sequence (GRCh37/ hg19) was used for genomic assembly.
The total number of read bases was 232,624,950 with a mean depth coverage of 0.08x. Signal duplication in the Xq28 region, which includes MECP2 and adjacent genes and spans approximately 525 kb, was observed in the patient (Fig. 1A). Duplication of MECP2 is associated with mental retardation, Lubs type (MIM 300260), an X-linked recessive neurodevelopmental disorder [10]. The final karyotype was identified as 46,XY.arr[hg19]Xq28(153,060,001-153,585,001)x2. To validate these results, a multiplex ligation-dependent probe amplification (MLPA) test was performed using SALSA MLPA P015 MECP2 probemix (MRC-Holland, Amsterdam, The Netherlands) according to the manufacturer’s instructions. MLPA test results revealed duplication of probe sites on all exons of MECP2, IRAK1, and FLNA in the patient (Fig. 1B). Family studies showed that his mother also carried the 525 kb duplication in the same region (Fig. 1C). The patient’s sister refused the genetic test; therefore, her carrier status could not be determined.
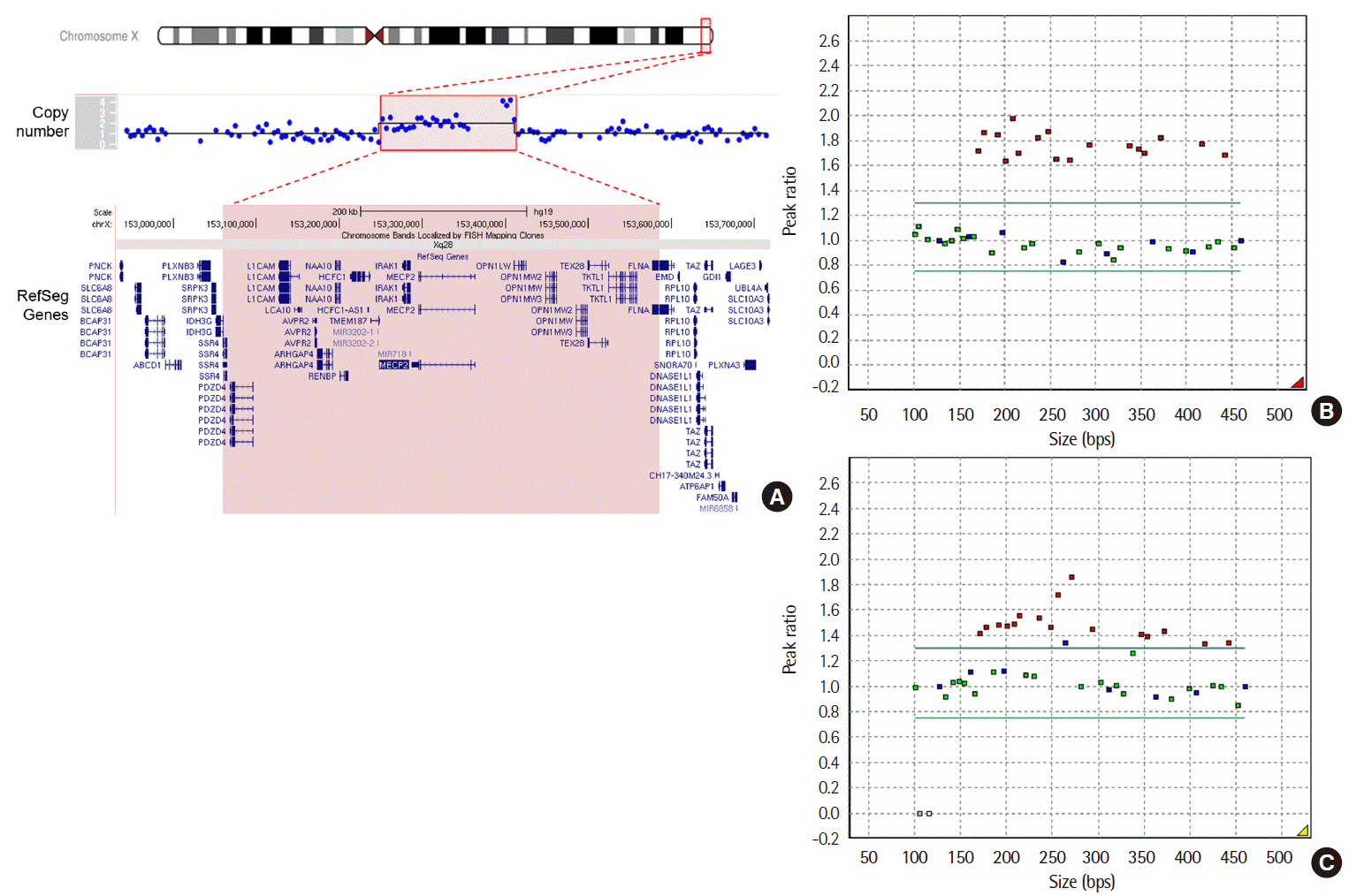 | Fig. 1Molecular investigations of a patient with intellectual disabilities, intractable epilepsy, and autistic features. (A) Low-depth whole-genome sequencing revealed a duplication of 525 kb at Xq28. The duplicated region (chrX:153,060,001-153,585,001, shown in red) contains MECP2 and several other genes. (B-C) multiplex ligation-dependent probe amplification analysis of MECP2 in the patient (B) and mother (C). The red squares represent duplications of MECP2 exons in each sample.
|
Chromosomal duplication in the Xq28 region, which includes MECP2, causes an X-linked neurodevelopmental disorder characterized by mental retardation, infantile hypotonia, mildly dysmorphic features, absent speech, autistic features, seizures, progressive spasticity, and recurrent infections [10]. The vast majority of affected males inherit the duplication from their mother, similar to our patient [10]. Although MECP2 is the primary gene responsible for the aforementioned neurological phenotypes [11], several previous studies suggest possible correlations between clinical phenotypes and the duplication of other genes [10, 12]. For instance, a previous study suggested that FLNA duplication may contribute to severe chronic intestinal pseudo-obstruction phenotypes seen in some patients with Xq28 duplication [12]. In addition, IRAK1 is a member of the toll-like receptor signaling pathway, suggesting the possible occurrence of immune system phenotypes as observed in patients [10]. To assess the genotype-phenotype correlation in Korean patients with MECP2 duplications, we compared the clinical features of our evaluated patient with previously reported cases (Table 1) [10, 13], and this analysis revealed shared core neurological features of MECP2 duplication syndrome, including constipation and recurrent infections.
Table 1
Comparison of clinical features of patients in this study and previous studies with MECP2duplications
| Phenotype | This study | Yon et al. (2017), first Korean cases [13] | % from Ramocki et al. (2010) [10] | |
|---|---|---|---|---|
|
|
|
|||
| Patient | Patient 1 | Patient 2 | ||
| Sex, current age | Male, 14 yr | Male, 11 yr | Male, 10 yr | NA |
| Duplication size (kb) | 525 | 411 | 411 | NA |
| Coordinates: first to last nucleotide (hg19) | 153,060,001:153,585,001 | 153,027,303:153,438,781 | 153,027,303:153,438,781 | NA |
| Infantile hypotonia | + | + | + | 92% |
| Mental retardation | + | + | + | 99% |
| Language development | Delayed | Delayed | Delayed | 88% |
| Autistic features/autism | + | + | + | 76% |
| Dysmorphic features | + | + | + | 100% |
| Epilepsy | + | + | + | 52% |
| Onset age | 48 months | 10 years | 10 years | |
| Type | Multiple types: focal clonic seizure, atonic seizure, and GTC | GTC | GTC | |
| Status epilepticus | + | ND | ND | ND |
| Treatment | valproic acid, oxcarbamazepine, and topiramate | oxcarbamazepine | valproate | ND |
| Brain MRI | ND | Normal | Normal | ND |
| Recurrent infections | + | + | + | 74% |
| Constipation | + | ND | ND | 76% |
![]()
Although the methods for CNV detection from WGS data have been available recently [3, 14], the cost of sequencing, the long turnaround times for deep-coverage WGS (>30x coverage using paired-end reads), and the heavy computational requirements for analysis have been major obstacles for the adaptation of WGS-based methods for cytogenetic applications. In this study, we identified a rare MECP2 duplication in a patient using a low-depth WGS strategy. Several studies have evaluated the low-depth NGS method for CNV detection. A previous study identified 71.8% (56/78) of subchromosomal abnormalities using 3.5 million reads [15], while a study conducted by Lo et al. [16] produced 64.5% (20/31) accuracy when 4-6 million reads were used to analyze samples with CNVs. The method used in the current study could not detect sequence variants or mosaicism due to the low read-depth (0.08x); however, a 525 kb duplication of MECP2 was accurately detected. The cost of this method is around 200 USD per sample and the turnaround time is within 1 week.
Developmental delays and intellectual disabilities occur in 1-3% of the general population [17]. Recent advances in genetic testing and the understanding of genetic disorders have led to changes in approaching diagnoses for children with unexplained developmental delays or intellectual disabilities [18]. In this study, we identified MECP2 duplication in a male patient with global developmental delays, intractable epilepsy, and autistic features. Our study revealed the potential use of the low-depth WGS to facilitate genetic diagnosis in clinical cytogenetics.
Go to : 
 XML Download
XML Download