

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Amyloidosis is a rare disease caused by extracellular deposition of amyloid, a fibrous substance derived from various proteins [1]. Amyloid light-chain (AL) amyloidosis occurs in 9 cases/million inhabitant/year in developed countries. The average age of diagnosed patients is 65 years and less than 10% of patients are under 50 [2]. Amyloidosis can affect all organs, including the brain, lungs, liver, heart, skin or kidneys. Invasion to the kidney is the most common, and more than 40% of patients progresses to end-stage renal failure after 3 years [3]. Both amyloid A (AA) and AL amyloidosis have been reported in many cases of recurrence after renal transplantation [4,5]. However, newly developed AL amyloidosis after renal transplantation is not common. We report a case of newly diagnosed AL amyloidosis in a kidney transplant recipient with focal segmental glomerulosclerosis (FSGS).
CASE REPORT
The study was approved by the Institutional Review Board of The Catholic University of Korea, Seoul St. Mary's Hospital (IRB No. KC19ZESE0140). Informed consent was not obtained as the patient had died at the time of writing this manuscript.
A 46-year-old female patient first presented on March 2015 with generalized edema and nephrotic syndrome. Kidney biopsy was done and she was diagnosed with FSGS. Reconfirmed kidney tissue was also diagnosed as FSGS. The patient was in good condition, but the renal function gradually worsened despite treatment. Afterwards, she developed to end-stage kidney failure and underwent kidney transplantation on March 2016. She received a living donor (sister) 4 antigen-matched kidney allograft. Blood group of patient and donor were all A positive. She had negative complement-dependent cytotoxicity and flow cytometric T-cell and B-cell cross-matches and panel reactive antibody was 0%.
Two years after renal transplantation, graft renal biopsy was performed with proteinuria and recurrent FSGS was confirmed by light microscopy. The patient was discharged after treatment with plasma exchange, but did not show improvement of proteinuria. One month after discharge, the deposits of amyloid fibrils on the electron microscope were confirmed and readmitted for further examination.
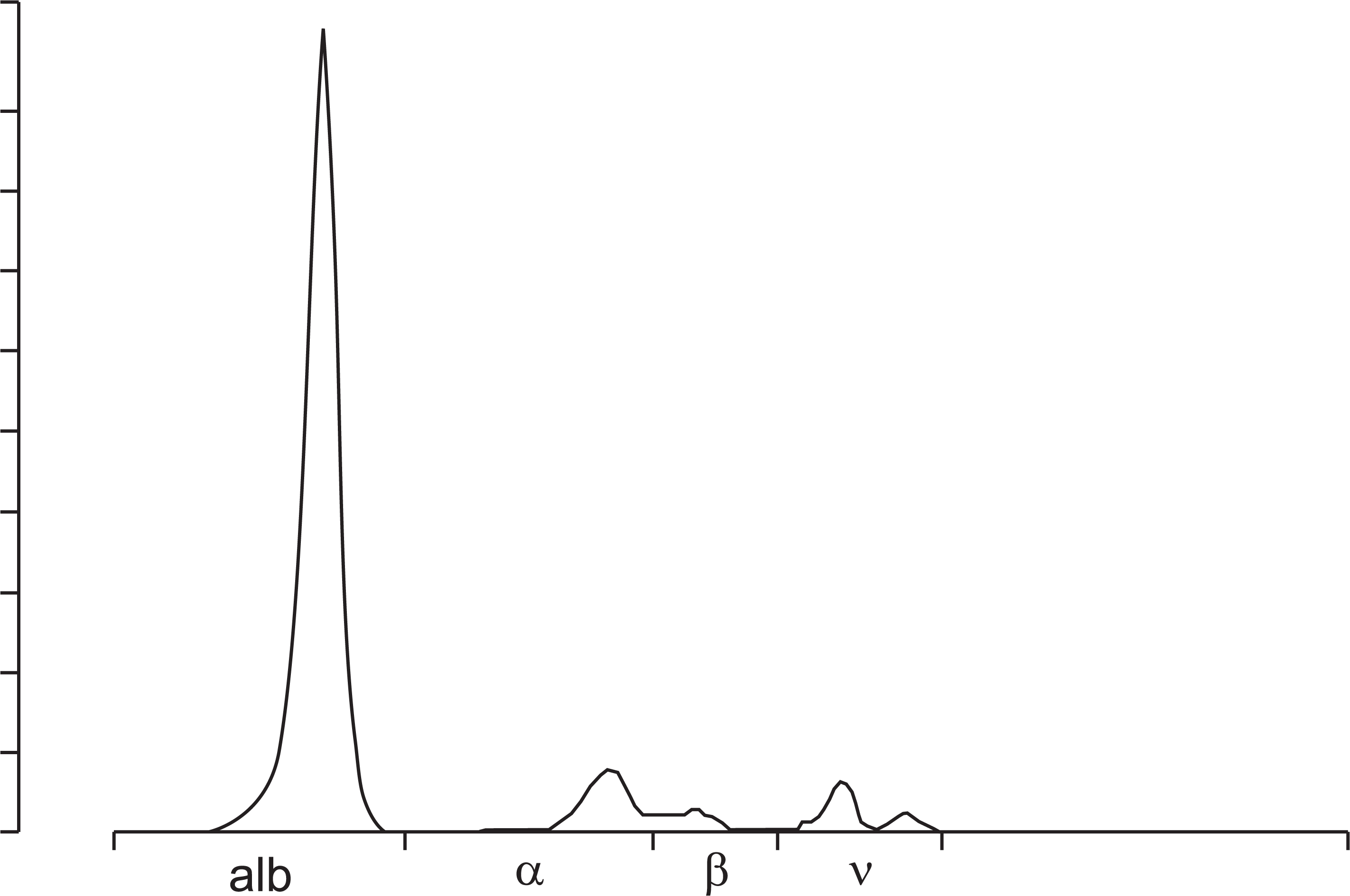
The patient had exertional dyspnea, general weakness, and visual field impairment. On examination, the blood pressure was 90/60 mmHg, the heart rate 20 beats per minute, the respiratory rate 20 breaths per minute, and the body temperature 36.5°C. Laboratory investigations showed white blood cells 7,150/mm3 (3,800–10,000/mm3), hemoglobin 11.2 g/dL (13.3–16.5 g/dL), and platelet 347,000/mm3 (140,000–400,000/mm3). The blood urea nitrogen was 31.3 mg/dL (9.0–23.0 mg/dL), creatinine 0.99 mg/dL (0.7–1.3 mg/dL), sodium 143 mEq/L (132–146 mEq), troponin-T 0.095 ng/mL (less than 0.014 ng/mL), and pro-BNP 4,395 pg/mL (0–249 pg/mL). On urinalysis, the urine protein/creatinine ratio was 8.23 and urine protein electrophoresis showed heavy albuminuria but no monoclonal protein (Fig. 1). In the serum free light chain assay, the kappa light chain was in the normal range of 6.80 mg/dL (3.30–19.40 mg/dL), but the lambda light chain showed abnormal results of 404.56 mg/dL (5.71–26.30 mg/dL). The ratio of kappa/lambda was abnormal result of 0.02 (0.26–1.65). Electrocardiogram showed low QRS voltage in limb leads. On echocardiography, the left ventricular ejection fraction was 68%, it showed a bilateral ventricular hypertrophy and a speckled pattern of myocardium typically seen in invasive cardiomyopathy, suggesting that amyloidosis was invaded into the heart. Analysis of bone marrow specimens showed an increased percentage of plasma cells (10%) and immunostaining revealed a positive reactivity for a lambda light chain.
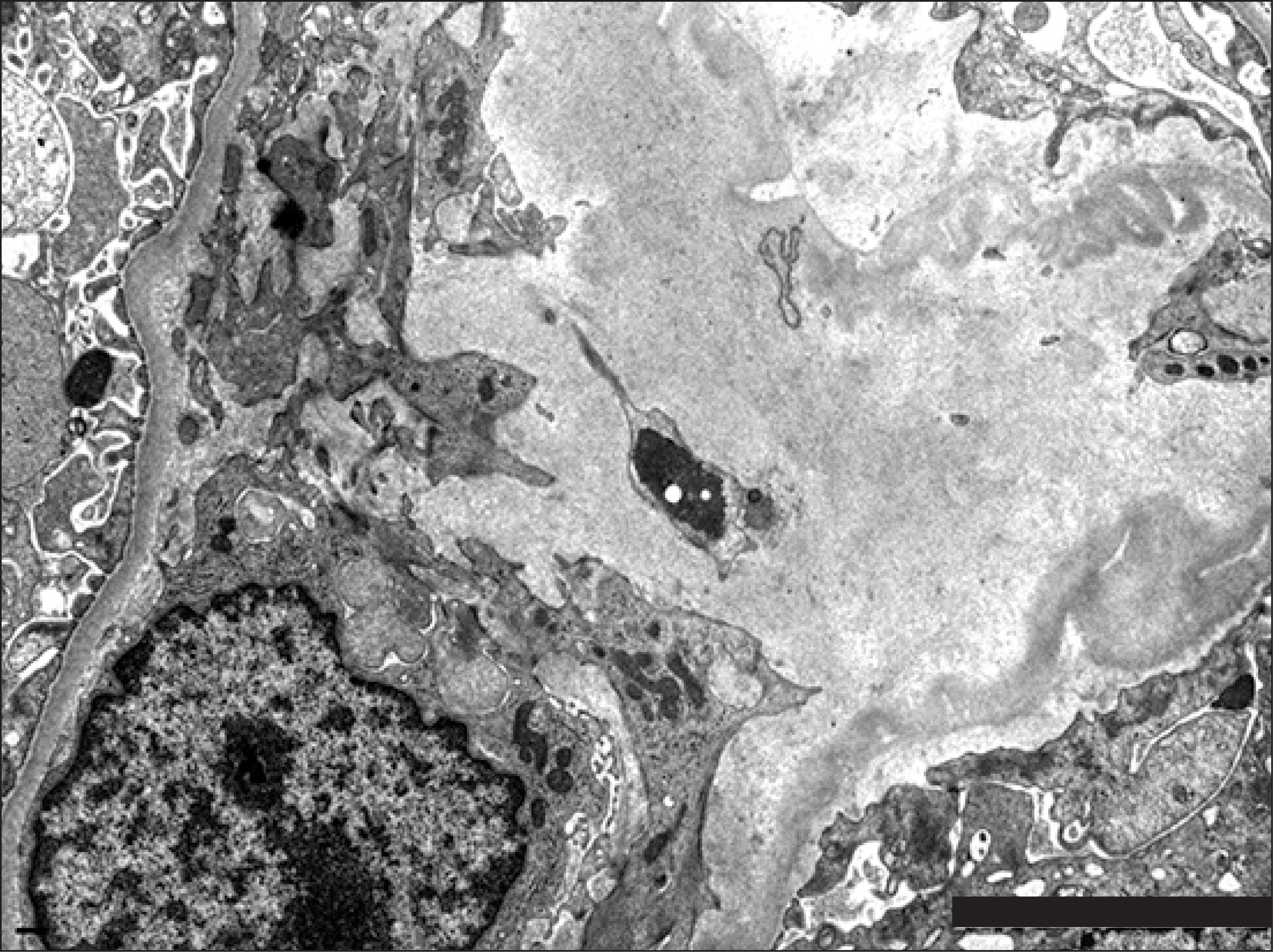
Graft kidney biopsy was performed and showed 11 glomeruli, two glomeruli displayed segmental sclerosis. A small amount of amorphous eosinophilic material deposition was locally invaded in the glomerular mesangium (Fig. 2). Immunofluorescence stain revealed small amount of mesangial deposition for lambda light chain (Fig. 3). Congo red staining showed characteristic apple-green color of amyloidosis and KMnO4 staining to differentiate between AA amyloidosis and AL amyloidosis showed positive finding. In addition, electron microscope showed small amount of haphazardly arranged non-branching fine fibrils (Fig. 4), which could be diagnosed as AL amyloidosis. The patient is currently planning chemotherapy and autologous bone marrow transplantation.
DISCUSSION
AL amyloidosis is a disease in which plasma cells fail to function properly and produce large amounts of free light chains and free light chains are deposited in various tissues. AL amyloidosis is often associated with one or more organs, especially when invading the heart, it is known that rapid diagnosis and treatment are important because it can proceed rapidly without proper treatment [6]. In 1998, Le et al. [7] first reported de novo amyloidosis after renal transplantation. In 2011, Qian et al. [8] reported four cases of newly developed AL amyloidosis after renal transplantation. In all cases, immunohistochemical staining of graft renal biopsy showed positive findings of congo red and lambda light chain. In Korea, newly diagnosed AL amyloidosis after renal transplantation is known only one case in 2016 by Hwang et al. (Table 1) [9].
The diagnosis of AL amyloidosis should satisfy all four criteria [10]. First, there should be systemic symptoms related to amyloid. Second, Congo-red staining should be positive in kidney biopsy. Third, there should be evidence that free light chains are associated with amyloidosis on immunohistochemical staining. Fourth, there must be evidence of abnormal proliferation of mononuclear cells. In this patient, immunohistochemical staining showed positive findings of congo red and lambda light chain deposit. Bone marrow examination showed 10% plasma cells and serum kappa/lambda ratio was abnormally low, which could lead to diagnosis of AL amyloidosis.
The main treatment for systemic AL amyloidosis is the combination therapy with chemotherapy and autologous bone marrow transplantation. The primary consideration in the treatment of AL amyloidosis is whether autologous bone marrow transplantation is possible. Autologous bone marrow transplantation is performed mainly in patients with low systemic risk. If pro-B-type natriuretic peptide <5,000 ng/L, troponin T <0.06 ng/mL, systolic blood pressure> 90 mmHg, age <65 years, and glomerular filtration rate >50 mL/min/1.73 m2 are satisfied, AL amyloidosis patients are treated with chemotherapy and autologous bone marrow transplantation. The combination of melphalan and dexamethasone has a significant survival rate in patients [11]. Because the patient of our case is in a low-risk group, the combination therapy with melphalan based chemotherapy and autologous bone marrow transplantation is being considered.
The patient had elevated serum BNP, low-voltage electrocardiogram, and invasive cardiomyopathy on echocardiography, indicating that amyloidosis is likely to have invaded the heart, but it is necessary to confirm it through myocardial biopsy. Unlike the previous cases, our case was initially diagnosed as recurrent FSGS on light microscopy and treated for recurrence, but the patient's symptoms and proteinuria did not improve. The patient was finally diagnosed as amyloidosis based on electron microscope finding 1 month after the initial diagnosis, which made it difficult in diagnosing AL amyloidosis. When the patient was admitted with increased proteinuria, AL amyloidosis would have been diagnosed quickly if additional tests for proteinuria were performed. Therefore, when the cause of chronic renal failure is FSGS, if proteinuria occurs after renal transplantation, recurrence of FSGS may be considered first. However, as in this case, we have experienced a need for differential diagnosis of renal diseases in which proteinuria may occur if the patient does not respond to treatment such as plasma exchange after renal transplantation.

 XML Download
XML Download