

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Congenital absence of portal vein (CAPV), also known as congenital extrahepatic portosystemic shunt (CEPS) was first described by John Abernethy in 1793 in London, England. It was a 10-month old girl who died of unknown reason. On postmortem examination, many congenital anomalies were found with dextrocardia, great vessel transpositions, polysplenia and portal vein bypass the liver and terminated into the inferior vena cava (IVC) at the renal vein level.1 Howard and Davenport2 referred the term “Abernethy malformation” for any extrahepatic congenital porto-caval shunt, this diversed visceral blood directly to IVC without passing the liver.
The classification of extrahepatic portal-caval shunts by Morgan-Superina in Table 13 was widely accepted.
CAPV once considered very rare congenital anomaly. Until 1997, Howard and Davenport2 reported 5 cases of congenital porto-caval shunt, including one end-to-side shunt which were the 13th of such cases in worldwide literature. Sokollik et al.4 (2013) resumed Pubmed reports and found 185 extrahepatic porto-caval shunt, with 103 (56%) type 1 and 82 (44%) type 2. Intrahepatic shunt were encountered in 131 patients.
Today, with advanced imaging techniques, the reported number of CAPV has been increasing, though most of these were case or serial report. Futhermore, mass screening for hypergalactosemia in neonates in Japan in the last decades accounted for the early and highest prevalence of CAPV in this country.5-8
The diagnosis is often incidental and made primarily by Doppler ultrasonography (US), but computed tomography (CT) or magnetic resonance (MR) are required to classify the shunt and to assess associate anomalies.
CASE
A 10-year old boy transferred to Vinmec Hospital (Hanoi, Vietnam) due to a huge mass on the right liver. His height of 137 cm, weight 24.6 kg, BMI 13.11 kg/m2. Medical history of congenital cardiac anomaly, diagnosed at 20 days after birth (atrial and ventricular septal defects; pulmonary hypertension up to 50 mmHg). During childhood he had to admit to hospital frequently (5-6 episodes per year) due to severe pneumonia. Under medical treatment and follow-up, all cardiac septal defects recovered spontaneously and pulmonary hypertension resolved. Since 4 years old, severe pneumonia became less frequent with 1-2 episodes per year. Skeletal anomaly particular noted with agenesis bilateral 5th toes.
On referral, his serum biochemical panel from previous hospital show intermittent disorders of liver function as in Table 2.
On admission at our hospital, he presented growth retardation and mild mental impairement, transient postprandial drowsiness, and loss of concentration. His height was in standard range, but his weight was 6.1 kg less than standard reference (comparable with 8 year-old boy) and was evaluated as Tanner stage 1. For mental problem, he had trouble to cope with his friends and stayed at class level 1 elementary school while his friends went to the 4th class. He can still remember simple things like parents name, his home address and so on; but got a long and hard for simple summation (within 10), can only spell but didn’t read well. On psychology examination, he was evaluated with Raven Colour Test: IQ 80 (ICD-10-CM: F70 - Mild intellectual disabilities) and Vineland Test 90 points.
Clinically, there were abdomen discomfort due to mild hepato-splenomegaly.
His total and direct bilirubin was 49.8 and 24.8 µmol/L (our hospital normal range was <21 and 5.1, respectively). AST 133.5 U/L and ALT 135.5 U/L (<35 and 35). GGT: 526 U/L (<55); ALP 676 U/L (30-120). NH3 90.6 µg/dl (18.7-86.9). AFP 2.29 (<9 ng/ml); PIVKA II 81.8 (0-40 mAU/ml). Liver virus panel and HIV negative. Platelet 326 G/L. INR (International normalized ratio) 1.04. PELD score: 2.8; Child Pugh Score 7 (Grade B). Cardiosonography found no more congenital defects, with EF 68%, without pulmonary hypertension.
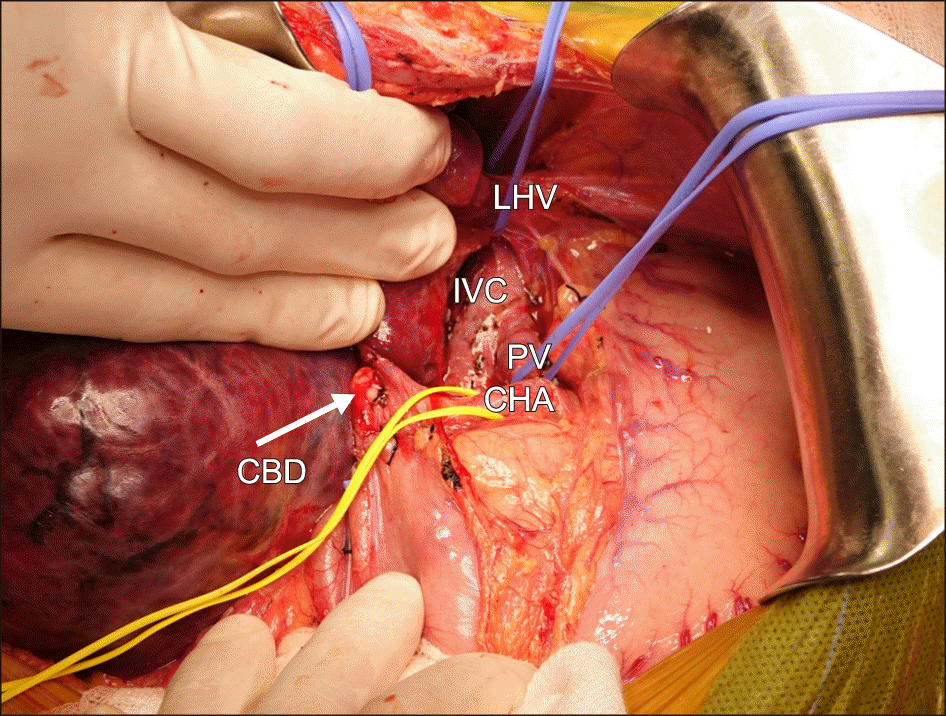
Living donor liver transplantation underwent using right lobe from his mother on 22 August 2018. Operation time was 430 minutes, anhepatic phase: 73 minutes. Warm ischemic time (graft out from cold preserve solution for RHV and portal vein reconstruction): 28 minutes; Cold ischemic time: 82 minutes. Portal vein clamping time: 16 minutes. Blood loss 550 ml; Red blood cell transfused: 350 ml. Graft weight: 549 gr; GRWR (Graft to Recipient Weight Ratio): 2,2%; G/SLV (Graft/Standard Liver Volume): 80%.
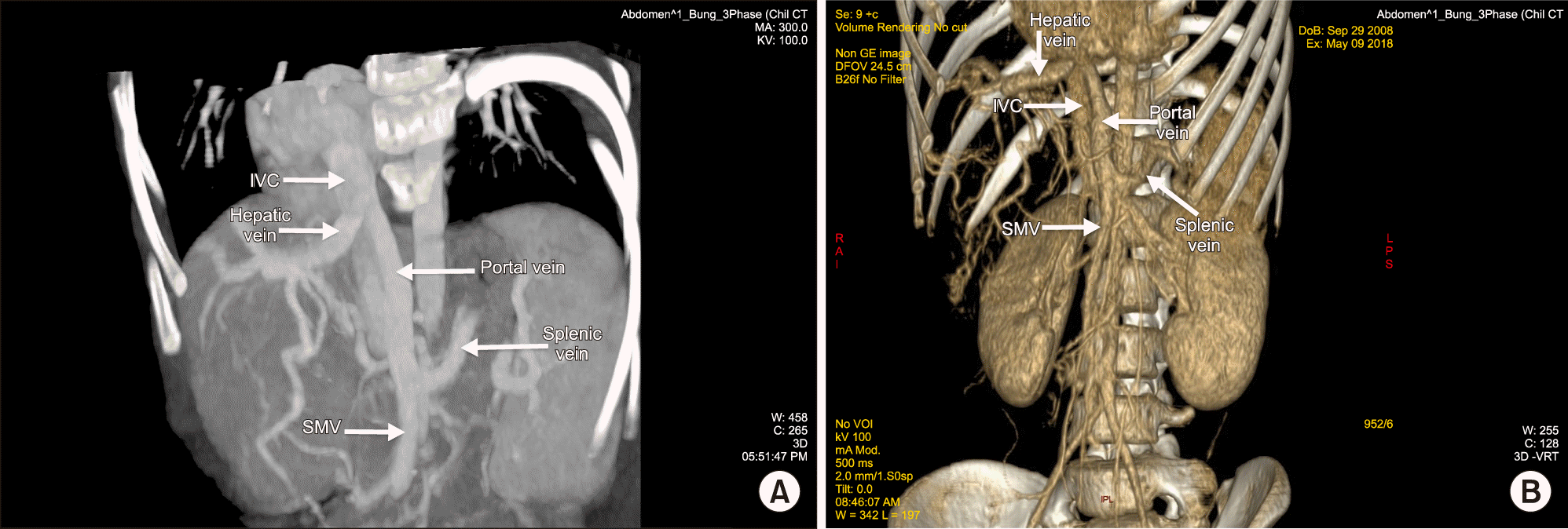
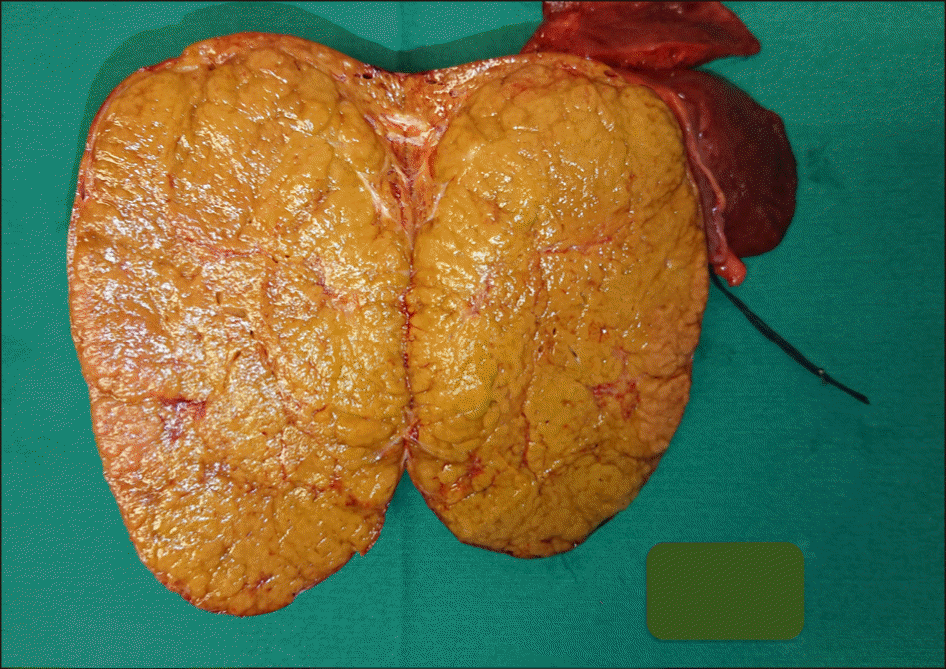
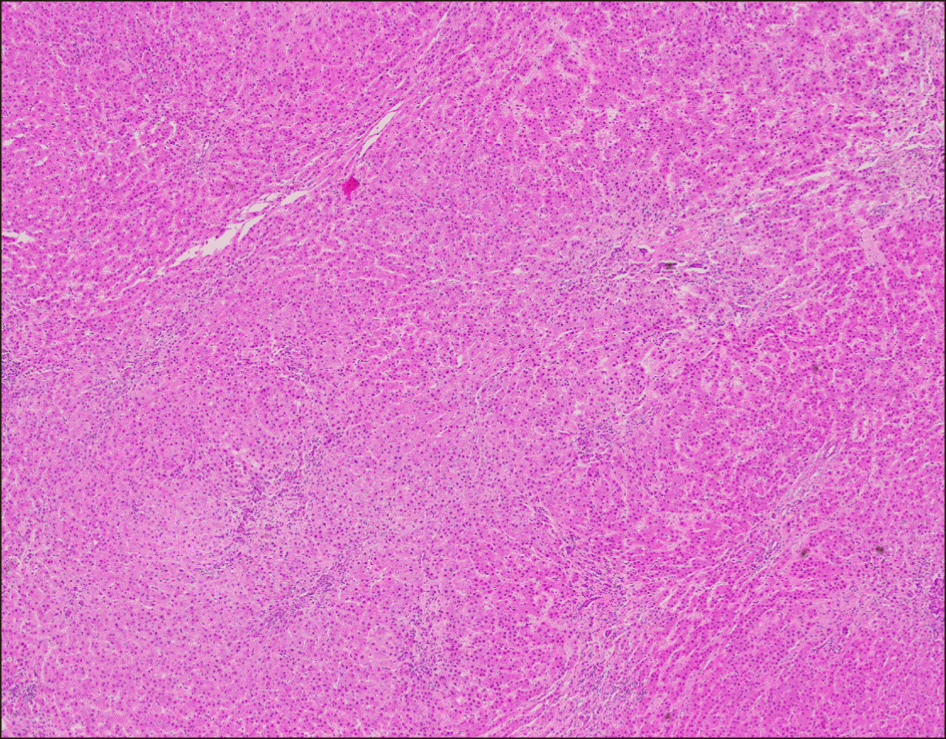
Intraoperative we found the native liver was almost dominated by a huge mass (mainly in the right liver). Mobilizing the whole liver from its ligaments, we confirmed that the patient had no right hepatic vein, the middle and left hepatic vein joined into a common trunk, draining into the IVC. The superior mesenteric vein confluenced with splenic vein before draining directly into the inferior vena cava (Abernethy anomaly type 1b), just 3 cm below and to the left of the draining site of hepatic vein complex into the IVC. The so-called portal vein measured of 10mm diameter and ran a 3 cm distance to the IVC (Figs. 1-3). Anatomopathology findings of the native liver showed chronic hepatitis with cirrhosis 4/6 Knodel-Ishak (Fig. 4).
The immunosuppressive inducted by basiliximab, then maintained with tacrolimus and mycophenolate.
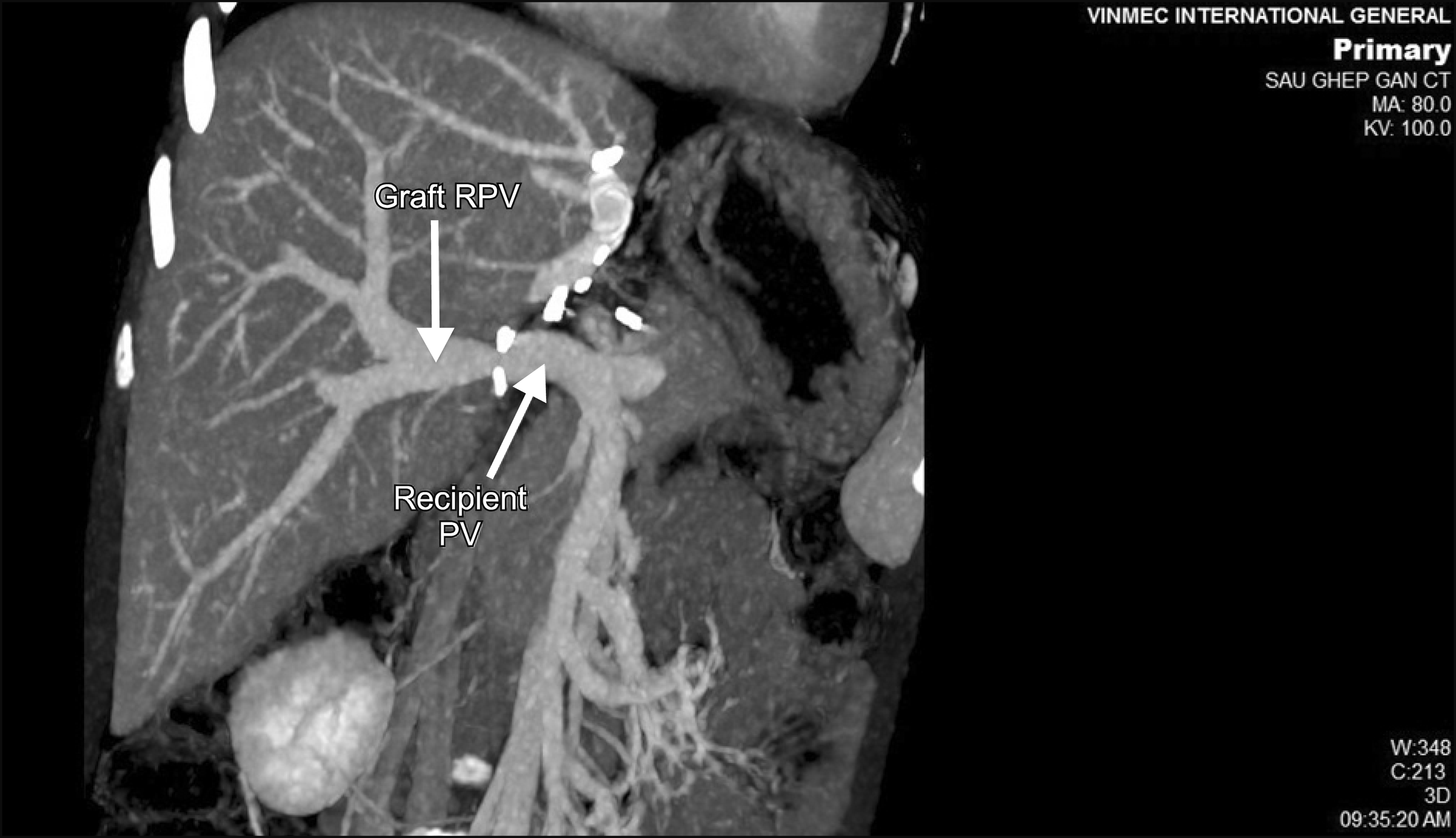
The post operative course was uneventful; the allograft function’s well with normalization of total bilirubin within 3 days, and AST/ALT down under 2N (upper normal value) within 9 days. CT Scan control 3 weeks postopratively showed all hepatic vessels patent with good flow (Fig. 5). Patient was discharged 28 days postoperatively. He was doing well at the follow-up 12 months later. His mentality was much improved according to his parents.
DISCUSSION
CAPV were diagnosed in children in 80% with age ranges from the prenatal period to a 64 years.5 The extrahepatic shunts were not significant difference between male and female proportion, however male gender were found in majority of intrahepatic shunts.4,7
Sakamoto et al.6 found in published literature 34 liver transplants related to CEPS worldwide to the year 2012. Most cases were diagnosed in childhood, except for 2 cases in adulthood (amongst 13 cases (38.2%) before one year old). Liver transplantation underwent at the age from 4 months to 45 years (mean 6.8 years). Male: female ratio was 15:19. The initial clues that lead to CEPS diagnosis were related to hepato-pulmonary syndrome or pulmonary hypertension in 9 cases (26.5%), hypergalactosemia at neonatal screening in 9 (26.5%). Six other cases (17.6%) related to biliary atresia, while hepatic encephalopathy (HE) including mild and transient symptoms, were met in 3 cases (8.8%). Altogether 23 cases (67.7%) were multi-anomalies (cardiac defects, polysplenia, biliary atresia, situs inversus…).6
Together with our case, worldwide literature also noted such concurrent anomalies in heart and skeletal system.9,10
This malformation presented with a wide range of clinical signs and symptoms related to the type of shunt and other concurrent anomalies. Hepatic encephalopathy syndrome (HES) in various degrees, mental retardation due to hyperammonemia, hyperbilirubinemia, hypergalactosemia; jaundice, fatigue, right upper-quadrant pain.6,11 Kobayashi et al.9 found only 13.2% CEPS related with HES. The decreased ammonia concentration and urease activity of the patient's faeces by intestinal flora were demonstrated. This acts as a homeostatic regulation, accounts for the fact that hepatic encephalopathy clinically uncommon or transient and with a mild degree.11-13 This comply with our patient who suffered mild mental defect and blood ammonia just slightly elevated.
The liver abnormal presentation often seen with focal nodular hyperplasia, nodular regenerative hyperplasia or hemangiomas. Kobayashi et al.9 reviewed 136 cases of CEPS showing that 43% (55 cases) presented with liver tumors, and most of them benign (83.6%).6,14 Respiratory symptoms usually seen as cyanosis, orthodeoxia platypnea associated with hepatopulmonary syndrome (HPS).5,15
Amongst 34 liver transplantations worldwidely reviewed by Sakamoto et al.6 related to CAPV, the indications due to HPS in 13 cases (38.2%), respiration complications in 11 (32.4%), liver mass in 5 (14.7%) (2 cases with hepatoblastome and 1 hepatocellular carcinoma), 4 biliary atresia and one persistent rectal bleeding. The grafts came from 15 brain death donors and 19 living donors.
Our indication for the child based on the huge mass in the right liver and start to show deterioration of liver function (elevation of liver enzymes) related to Abernethy malformation type 1b.
Liver tumor in the CAPV is very common and mostly benign. Although hepatocellular carcinoma were reported by some authors, liver biopsy was not recommended routinely, though it helps clearify the malignancy as well as reveal the existence of small portal venules in the portal triads.14
The current treatment guideline is not standardized due to scattered data and various studies used different terminology, some equating the Abernethy malformation of any type of CAPV. However most of authors consider liver transplantation for uncontrol hyperammonemia, or HES or liver masses.5 Treatment options also depends on the type of porto-caval shunt. Liver transplantation is the most effective and practical for type 1 CAPV, while in type 2, cure can be ensured by surgical closure or interventional radiology (coil embolization or catheter-device closure).6,14 With potential risk of developing pulmonary hypertension and severe HPS (which preclude LT later) Brasoveanu as cited from Soejima et al.5 proposed prophylactic LT in children with CAPV.
Due to the lack of portal blood flow (accounts for 80% in normal circumstancy), it is particular finding that hepatic artery dilated in CAPV for compensation.1,5,16 In our case, the graft came from an adult (right hepatic artery diameter of 2 mm) to a child (common hepatic artery diameter of 2.3 mm), we successfully reconstructed the hepatic artery by manual without microscope.
Portal vein reconstruction could be extremely difficult due to the short portal conduit in some cases that require venous interposition graft.5 However in our patient the mesenterico-splenic confluence runs quite a long way enough to make a simple anastomosis (length of 3 cm).
CAPV was originaly not associated with portal hypertension, therefore there was no collateral circulation. This leads to intraoperative risk of important intestinal congestion during portal clamping and anastomotic performance.5,14,17 Sibuleski et al.18 experienced one case of intraoperative non-trauma splenic rupture due to abruptly portal hypertension while clamping the portal vein in liver transplantation. In the present case, we hold the portal phase clamping to the very last moment, just when finished the hepatic vein anastomosis and ready for a portal vein reconstruction. This helped minimize portal clamping time (to 28 minutes) without consequence on vesceral perfusion. There are other reports of portal clamping in liver transplant due to CAPV duration in 15-17 minutes without significant consequence on intestinal edema. However in theory, it should be borne in mind that mesenterical pressure might be elevated due to resistence of the new liver graft that could affect the intestinal perfusion postoperatively.3,5
Liver transplantation was a justified indication for congenital absence of the portal vein type 1, presented with huge liver parenchyme hyperplasia and liver failure. Living- donor liver transplantation was effective and practical in the scarcity of donor organ.
 XML Download
XML Download