

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Type 2 diabetes mellitus (T2DM) is strikingly increasing globally and leads not only to the threatened health of individuals but to the economic burdens of society [1]. Although dysfunction of many organs contributes to hyperglycemia, pancreatic β-cell dysfunction plays a key role in the development of diabetes. Thus, the preservation of β-cell is an essential component of the treatment strategy for T2DM [2].
When β-cell are exposed to chronically excess nutrients, insulin secretion increases initially but eventually fails to compensate for insulin secretion and loses their identity. Although the cause of the metabolic deterioration is unknown, several hypotheses have been proposed, including mitochondrial dysfunction, oxidative stress, endoplasmic reticulum (ER) stress, hyperglycemia (glucotoxicity), dyslipidemia (lipotoxicity), and the combination of both (glucolipotoxicity). The chronic hyperglycemia and hyperlipidemia cause combined, detrimental effects defined as glucolipotoxicity on β-cell function and survival. Though free fatty acids (FFA) stimulate insulin secretion, chronically elevated FFA impairs pancreatic β-cell function in vitro and in vivo, which leads to the induction of lipotoxicity. FFAs move into cells through a passive concentration-dependent diffusion, and it has been reported that there are active transport systems to enhance FFA uptake. Fatty acid translocase cluster determinant 36 (CD36), which is part of the FFA transporter system, has been identified in several tissues such as muscle, liver, and insulin-producing cells. Several studies have reported that induction of CD36 increases uptake of FFA in several cells linked to insulin resistance in diabetes, suggest that CD36 may play a pathophysiological event responsible for β-cell dysfunction and failure in T2DM. However, we do not currently know the regulating mechanism and pathophysiological role of CD36 and its upstream and downstream targets on glucolipotoxicity in pancreatic β-cells. In this review, we discuss the involvement of CD36 in hyperglycemia and hyperlipidemia (ceramide)-induced β-cell dysfunction along with the clinical studies on association between CD36 and metabolic disorders.
Go to : 
CD36 STRUCTURE AND FUNCTIONAL REGULATION
CD36 is a scavenger receptor present on various types of cells, including pancreatic β-cell, α-cells and contributes to lipid accumulation and in the progression of metabolic dysfunction. The human CD36 gene is located on chromosome 7 (7q11.2) and consists of 15 exons with a single peptide chain of 472 amino acids and has a predicted molecular mass of 53 kDa [3]. Analysis of amino acid sequence of CD36 predicts a two transmembrane domains and two short cytoplasmic tails at the N- and C-terminal result in a hairpin configuration (Fig. 1) [4]. The extracellular loop contains a large hydrophobic cavity responsible for the recognition of oxidized low-density lipoproteins (OX-LDL), advanced glycated end products, cholesterol, and fatty acids [56]. This binding site also contains multiple glycosylation sites and three disulfide bridges essential for the intracellular processing of the protein and its correct deliver and targeting from the extracellular space to the outer leaflet of the plasma membrane [7]. Additionally, the N- and C-terminal tails contain cysteine residues that palmitoylated to anchor the protein to the membrane bilayer. In detail, palmitoylation is a reversible enzymatic process, which requires palmitoyl-transferases (PATs) and palmitoyl-protein thioesterases (PPTs) for palmitoylation and depalmitoylation, respectively. A recent study by Wang et al. [8] demonstrate that Asp-His-His-Cys (DHHC) motif-containing palmitoyl acyltransferases, DHHC4 and DHHC5, regulate the palmitoylation of CD36 in regulating fatty acid uptake activity in adipose tissues. CD36 palmitoylation and localization of CD36 on the plasma membrane of hepatocytes are increased in patients with non-alcoholic steatohepatitis as well as livers in mice with non-alcoholic steatohepatitis. Inhibition of palmitoylation of CD36 protects mice from non-alcoholic steatohepatitis and inflammatory response [9]. Notably, palmitoylation might regulate the glycosylation of CD36 and glycosylation of CD36 is necessary for proper folding and trafficking to the plasma membrane, but does not affect ligand binding [1011]. In contrast to palmitoylation, O-GlcNAcylation of CD36 induces CD36 translocation to the sarcolemma and subsequently enhances myocellular fatty acid uptake [12]. On the other hand, increased O-GlcNAcylation of CD36 has also been linked to coordinate reprogramming of cardiac fatty acid and glucose metabolism, thereby maintaining cardiac energetics during acute or chronic stresses [13].
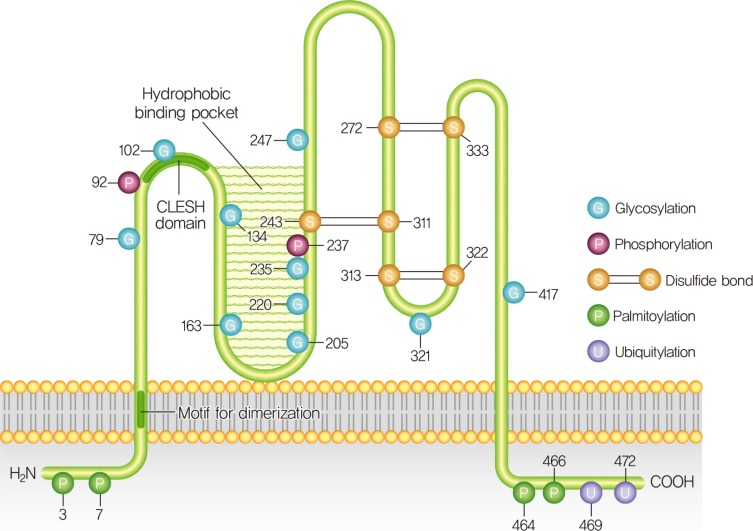 | Fig. 1Cluster determinant 36 (CD36) structure and post-translational modifications. CD36 has two transmembrane domains and two small cytoplasmic tails that contain four palmitoylation sites. The C-terminus contains two ubiquitylation sites and the N-terminal transmembrane domain contains two motifs (G12xxxG16xxxA20 and A20xxG23) that are responsible for dimerization. The large extracellular loop contains 10 N-linked glycosylation sites and two phosphorylation sites. There are two binding entrance in extracellular domain: the one is hydrophobic pocket bind with a variety of ligands, and the other is for fatty acid transport. CLESH, CD36, LIMP-2, Emp sequence homology.
|
CD36 has two phosphorylation sites at Thr92 and Ser237, both of which are within the extracellular loop linked to CD36 function, possibly through the modulation of ligand binding [1415]. CD36 has four acetylation sites, Lys52, Lys166, Lys231, and Lys403, and the biological role of these acetylations for CD36 expression and/or functioning have not yet been known [16]. It has been known that CD36 has two polyubiquitylation sites in the C terminus at Lys469 and Lys472 and ubiquitination of CD36 did not affect the relative CD36 distribution between the intracellular storage compartments and cell surface [17]. It was observed that CD36 can be monoubiquitinated by Parkin, an E3 ubiquitin-protein ligase leading to an increase in its stability at the plasma membrane [18]. Parkin-knockout mice on a high-fat diet had reduced hepatic CD36 levels resulted in abnormalities of fatty acid utilization. Taken together, CD36 could be targeted by different, yet unidentified E3 ligases in different tissues underscore the notion that CD36 ubiquitination are strictly tissue specific that warrants further investigation.
Go to :
CD36 ROLE IN PANCREATIC β-CELL DYSFUNCTION DURING HYPERGLYCEMIA
CD36 has been shown to participate in multiple homeostatic and pathological processes. This functional diversity is attributable to the multiple and distinct binding sites present on CD36. In β-cells, CD36 was found in the plasma membrane as well as intracellular and co-localized with insulin granules [19]. Several studies have shown that fatty acids can induce β-cell death via apoptosis, which was potentiated by glucose [2021]. Our lab has been interested to study the function of CD36 as a scavenger receptor in pancreatic β-cell and found that CD36 influences glucotoxicity dysfunction by increasing the influx of FFAs into pancreatic β-cells [22]. Moreover, forced expression of CD36 in β-cells increase the uptake of fatty acids and leads to metabolic and functional alterations [23]. CD36 was shown to traffic between the cell surface and intracellular compartments in a vesicle-mediated process [24]. However, little was known about the expression and trafficking of CD36 in the β-cell cells. We found that Rac1, a small guanosine triphosphate (GTP)–bound protein belonging to the Rho family had increased glucose mediated CD36 expression on the membrane surface in pancreatic β-cells which promoted signal transduction [25]. On the other hand, increasing evidence suggests that chronic elevation of glucose leads to the generation of reactive oxygen species (ROS), resulting in increased oxidative stress in β-cells [262728]. Elevated levels of glucose activate Rac1, which subsequently increase oxidants such as H2O2 via activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase [29]. Recently, it has been shown that CD36 deficiency attenuates obesity associated oxidative stress in the heart through reduced NADPH oxidase activity [30]. Although evidence of a direct interaction between NADPH oxidase and CD36 is still not known, we found that inhibitors of each target can abrogate the downstream signaling damage in response to high glucose. Moreover, it should be noted that changing the balance of antioxidant enzymes and increasing ROS production can alter susceptibility to dysfunction by the activation of stress kinases. Of relevance to redox signaling, oxidized lipids induced CD36 downregulates the redox-sensitive nuclear factor Nrf2 via Src family kinase member Fyn mediated signaling in murine vascular smooth muscle cells [31]. In addition, CD36 signaling in response to scavenger ligands leads to recruitment and activation of Src family non receptor protein tyrosine kinases and serine/threonine kinases of the mitogen-activated protein kinase (MAPK) family, such as, Lyn and c-Jun N-terminal kinases (JNK) in macrophages and platelets, whereas Fyn and p38 are the primary mediators of endothelial cell [323334]. Considering this, we found that high concentrations of glucose induced JNK and P38MAPK activation while Rac1 inhibition blocked this activation. In contrast with most other cell types, β-cells have relatively low levels of free-radical-detoxifying enzymes [3536]. Based on these, pharmacological inhibition of CD36 efficiently reduces downstream signaling pathways linked to high glucose and its subsequent toxicity. With this functional characterization, we hypothesize that CD36-mediated enhancement of oxidant stress contributes to pancreatic β-cell dysfunction. Given that CD36 expression is related to a diverse number of pathological functions, the identification of molecules that can modulate CD36 expression in response to high glucose requires further study in the pancreatic β-cells.
Go to :
CD36 ON LIPOTOXICITY MEDIATED β-CELL DYSFUNCTION
Accumulating evidence suggests that glucolipotoxicity contributes to β-cell dysfunction during the development of T2DM. Chronic exposure of β-cells to supraphysiological levels of glucose or FFAs has been shown to be cytotoxic and causes β-cell dysfunction and failure [373839]. Briaud et al. [40] have provided evidence that lipotoxicity occurs in the presence of concomitantly elevated levels of glucose. Several mechanisms have been proposed for glucolipotoxicity induced β-cell dysfunction and failure, such as increased ROS, ceramide, and nitric oxide levels, and mitochondrial perturbations [414243]. Recent studies suggest that hyperglycemia-induced CD36 expression mediates cellular dysfunction in various tissues as well as in the pancreatic β-cells [22444546]. As CD36 modulates cellular FFA transfer, studies with the Zucker diabetic fatty rat, an obesity induced diabetic animal model, indicate that elevated levels of FFA cause ceramide accumulation which destroys the β-cells by apoptosis [47]. Numerous studies have reported higher plasma levels of ceramides in patients with obesity and T2DM, suggesting that these atypical ceramides may serve as biomarkers for the diagnosis and treatment of obesity and diabetes [484950]. Based on its role in FFA uptake, our lab also showed that CD36 promotes ceramide induced β-cell functional defect through Vav family guanine nucleotide exchange factors (GEF). The GEF activity of Vav proteins is tightly regulated by Src-mediated tyrosine phosphorylation and concomitantly elevated various metabolic pathways [51]. We also showed that CD36 signaling leads to Src mediated Vav tyrosine phosphorylation and promotes a Rac1-NADPH oxidase (NOX) signaling pathway that generates ROS. In addition, evidences suggest that the abnormal Src signaling impairs insulin secretion and induces insulin resistance through saturated fatty acids in T2DM [52]. We also found that CD36 knockdown of expression by small interfering RNA (siRNA) or inhibition of Src activity, significantly blocked ceramide induced Rac1-NOX and inhibited ROS formation [53]. However, the mechanism behind this CD36 and Src kinase function is not known, but Holzer et al. [54] demonstrated that saturated fatty acids stimulate Src into membrane micro domains contributes to stress signaling activation. In addition, FFAs mediates Src-dependent phosphorylation of Vav and the subsequent engagement of Rac1-NOX signaling coordinates JNK activation contributes to insulin resistance, obesity and the production of inflammatory cytokines [5556]. Likewise, we also pinpoint the underlying mechanisms for CD36 signaling causes JNK activation as part of a generalized stress response by ceramide. The mechanism by which CD36-JNK interlink promotes β-cell damage is not completely understood.
p66Shc, a 66-kDa Src collagen homologue (Shc) adaptor protein, is implicated in both sensing and activation of cellular oxidative stress and the consequent induction of apoptosis [57]. It was recently shown that p66Shc mediates lipotoxicity-induced apoptosis in pancreatic β-cells, suggesting that p66Shc could sense the impaired metabolic changes in diabetes and promote cellular dysfunction [58]. Earlier studies have suggested that JNK-dependent p66Shc serine36 phosphorylation leads to ROS production and cell death [59]. Importantly, ceramide treatment showed that JNK and p66Shc were activated in a CD36-dependent manner. These signaling events subsequently promote translocation of p66Shc to mitochondria to drive reactive oxygen production. These then further promote activation of oxidized PRDX3 accumulation in the mitochondria would favor mitochondrial permeability transition pore (MPTP) opening and indeed mitochondrial swelling [53]. There are evidences pointing to the hyperoxidation of peroxiredoxin-3 within the mitochondrial compartment, ultimately resulting in the oxidation of the peroxidase cysteine to sulfonic acid (peroxiredoxin-SO3) [60]. The expression of peroxiredoxin-3 was restricted to β-cells in the pancreatic tissue, wherein it protects the β-cells against cytokine-induced damage. This reaction is redox-dependent and the reduced active form of peroxiredoxin-3 is regulated by the thioredoxin-thioredoxin reductase system. However, ceramide treatment decreased the expression of thioredoxin in pancreatic β-cells. We have shown that thioredoxin-interacting protein (TXNIP) translocates to the mitochondria and inhibits the antioxidative protein thioredoxin in response to ceramide [61]. This observation suggests the possibility that CD36-mediated effects are associated with nuclear factor κB dependent TXNIP induction. The inhibition of CD36 resulted in the restoration of mitochondrial thioredoxin via enhanced activity of thioredoxin reductase, a critical regulator of the active form of thioredoxin. The multiple pathways activated by CD36 suggest that further studies are in need to delineate the exact CD36 signaling pathways induced by ceramide in β-cell failure (Fig. 2).
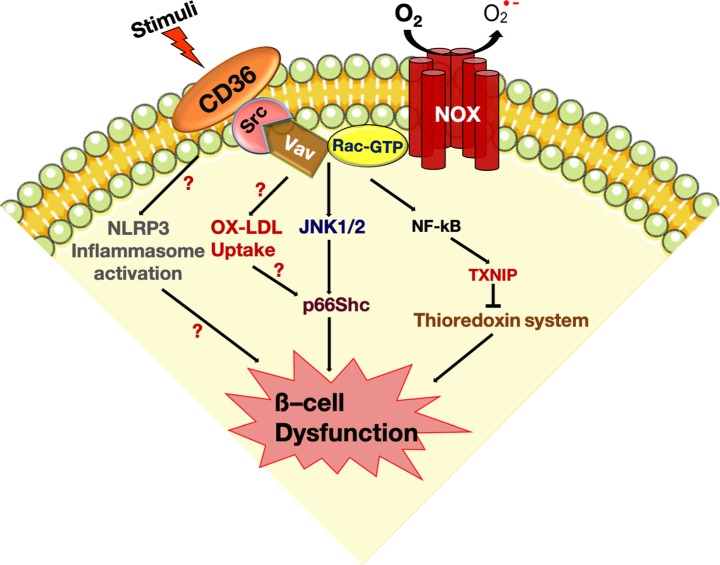 | Fig. 2Cluster determinant 36 (CD36)-induced signal transduction and damage in pancreatic β-cells, and its mechanisms. Binding of a variety of ligands to CD36 on the plasma membrane initiates the assembly of a redoxosome (Src-Rac1-nicotinamide adenine dinucleotide phosphate [NADPH] oxidase) complex. Redoxosome activation induces c-Jun N-terminal kinases (JNK) activation, thereby leading to p66Shc mediated mitochondrial peroxiredoxin-3 oxidation via thioredoxin-2 downregulation. Moreover, this complex activates nuclear factor κB (NF-κB) which in turn contributes to the impaired mitochondrial defense leads to pancreatic β-cell failure. Besides future studies are warranted to examine the relationship between CD36 and nucleotide-binding domain leucine-rich repeat (NLR) and pyrin domain containing protein 3 (NLRP3) inflammasome activation in the pancreatic β-cell dysfunction and failure. Activation of CD36 signaling by oxidized low-density lipoprotein (OX-LDL) initiates the activation of the signalosome complex and inflammatory programs such as the production of reactive oxygen species (ROS) and p66Shc contribute to the pathogenic pathway that links to β-cell dysfunction and failure. GTP, guanosine triphosphate; NOX, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase; TXNIP, thioredoxin-interacting protein.
|
Go to :
ROLE OF OX-LDL AND CD36 LINKS IN PANCREATIC β-CELL DYSFUNCTION
In addition to these proposed mechanisms, substantial evidence supports the central role of CD36 as a signaling hub to several signaling pathways [62]. As described above, it has been shown that OX-LDL induces β-cell dysfunction via ROS and radical lipid hydroperoxides [6364]. Moreover, prolonged incubation of human and rat islets to OX-LDL particles lead to JNK activation and downstream proapoptotic signaling. In addition, the blockade of OX-LDL-induced activation of JNK using a JNK inhibitor protected β-cells from the effects of OX-LDL [65]. However, the downstream mechanism by which JNK leads to apoptosis is not clear, and this may be assumable that crosslink between the OX-LDL and CD36 may determine cellular commitment to apoptosis through JNK activation. So, further investigation is needed to clarify these expected findings. Recent evidence suggests that ER stress was linked to oxidative stress in OX-LDL induced β-cell dysfunction and death [66]. In this context, we also showed that CD36 expression was induced in high glucose and thapsigargin-treated β-cells. Further, inhibition of CD36 with sulfo-N-succinimidyl oleate (SSO) or siRNA blocked high glucose induced ER stress markers and β-cell damage [67]. In addition, OX-LDL mediate JNK-dependent phosphorylation of p66Shc in endothelial cells contributes to oxidative stress and the atherogenic progression [68]. A recent study by Vandanmagsar et al. [69] reported that lipotoxicity associated increases in intracellular ceramide induce caspase-1 cleavage in macrophages and adipose tissue through nucleotide-binding domain leucine-rich repeat (NLR) and pyrin domain containing protein 3 (NLRP3) inflammasome activation. In pancreatic β-cells, TXNIP indeed has a role in producing IL-1β through NLRP3 inflammasome activation under ER stress [70]. Moreover, a recent study by Sheedy et al. [71] showed that an early CD36-mediated pathogenic inflammasome activation links cholesterol accumulation in the chronic inflammatory process of atherosclerosis. So, future studies are warranted that examine the relationship between CD36 and NLRP3 inflammasome activation in the pancreatic β-cell dysfunction and failure.
Go to :
CLINICAL STUDIES ON ASSOCIATION BETWEEN CD36 AND METABOLIC DISORDERS
Since CD36 is responsible for the transport of fatty acids, clinical studies have conducted to elucidated the role of CD36 as a biomarker or therapeutic target for metabolic disease in humans. Soluble CD36 (sCD36) is a non-cell bound CD36 which has been identified in human plasma and indirectly reflects CD36 expressed in tissues [72], and sCD36 has been shown to be associated with insulin resistance, carotid atherosclerosis, and fatty liver [73] in non-diabetic participants. Clinical studies about the association between sCD36 and cardiometabolic disorders are summarized in Table 1. Handberg et al. [74] reported for the first time that plasma sCD36 level was highly correlated with insulin resistance and body mass index (BMI) in patients with T2DM. Whereas sCD36 was also increased in polycystic ovary syndrome patients, pioglitazone treatment significantly reduced sCD36 levels with improving insulin sensitivity [75]. A recent community-based cohort study also proven that higher sCD36 is associated with adiposity (both subcutaneous and visceral) and non-alcoholic fatty liver disease with inflammation [76].
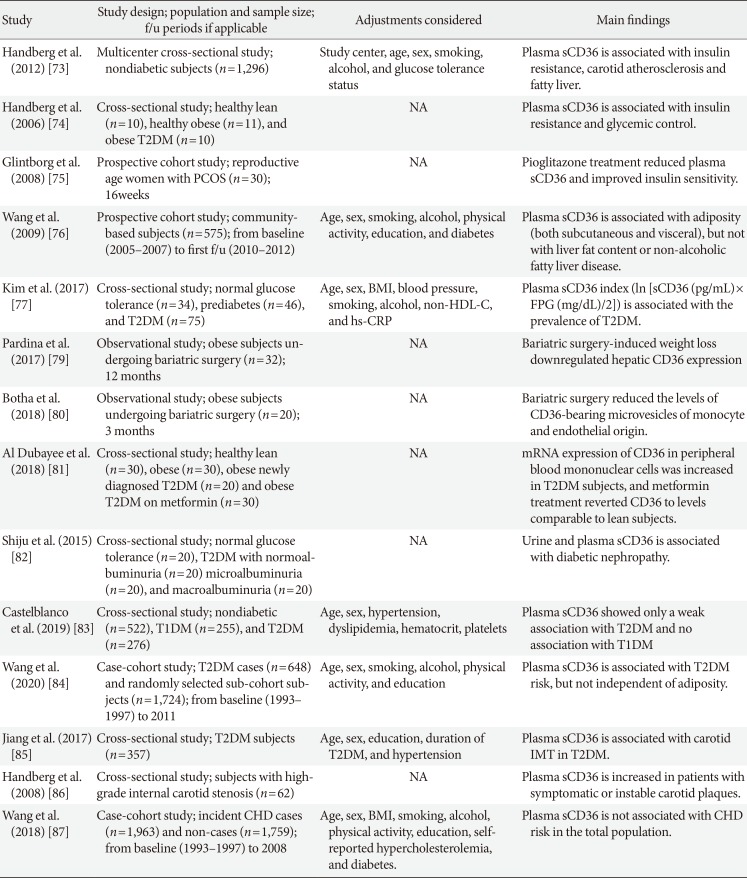
Table 1
Clinical studies of the association between sCD36 and cardio-metabolic disorders
| Study | Study design; population and sample size; f/u periods if applicable | Adjustments considered | Main findings |
|---|---|---|---|
| Handberg et al. (2012) [73] | Multicenter cross-sectional study; nondiabetic subjects (n=1,296) | Study center, age, sex, smoking, alcohol, and glucose tolerance status | Plasma sCD36 is associated with insulin resistance, carotid atherosclerosis and fatty liver. |
| Handberg et al. (2006) [74] | Cross-sectional study; healthy lean (n=10), healthy obese (n=11), and obese T2DM (n=10) | NA | Plasma sCD36 is associated with insulin resistance and glycemic control. |
| Glintborg et al. (2008) [75] | Prospective cohort study; reproductive age women with PCOS (n=30); 16weeks | NA | Pioglitazone treatment reduced plasma sCD36 and improved insulin sensitivity. |
| Wang et al. (2009) [76] | Prospective cohort study; community-based subjects (n=575); from baseline (2005–2007) to first f/u (2010–2012) | Age, sex, smoking, alcohol, physical activity, education, and diabetes | Plasma sCD36 is associated with adiposity (both subcutaneous and visceral), but not with liver fat content or non-alcoholic fatty liver disease. |
| Kim et al. (2017) [77] | Cross-sectional study; normal glucose tolerance (n=34), prediabetes (n=46), and T2DM (n=75) | Age, sex, BMI, blood pressure, smoking, alcohol, non-HDL-C, and hs-CRP | Plasma sCD36 index (ln [sCD36 (pg/mL)× FPG (mg/dL)/2]) is associated with the prevalence of T2DM. |
| Pardina et al. (2017) [79] | Observational study; obese subjects undergoing bariatric surgery (n=32); 12 months | NA | Bariatric surgery-induced weight loss downregulated hepatic CD36 expression |
| Botha et al. (2018) [80] | Observational study; obese subjects undergoing bariatric surgery (n=20); 3 months | NA | Bariatric surgery reduced the levels of CD36-bearing microvesicles of monocyte and endothelial origin. |
| Al Dubayee et al. (2018) [81] | Cross-sectional study; healthy lean (n=30), obese (n=30), obese newly diagnosed T2DM (n=20) and obese T2DM on metformin (n=30) | NA | mRNA expression of CD36 in peripheral blood mononuclear cells was increased in T2DM subjects, and metformin treatment reverted CD36 to levels comparable to lean subjects. |
| Shiju et al. (2015) [82] | Cross-sectional study; normal glucose tolerance (n=20), T2DM with normoal-buminuria (n=20) microalbuminuria (n=20), and macroalbuminuria (n=20) | NA | Urine and plasma sCD36 is associated with diabetic nephropathy. |
| Castelblanco et al. (2019) [83] | Cross-sectional study; nondiabetic (n=522), T1DM (n=255), and T2DM (n=276) | Age, sex, hypertension, dyslipidemia, hematocrit, platelets | Plasma sCD36 showed only a weak association with T2DM and no association with T1DM |
| Wang et al. (2020) [84] | Case-cohort study; T2DM cases (n=648) and randomly selected sub-cohort subjects (n=1,724); from baseline (1993– 1997) to 2011 | Age, sex, smoking, alcohol, physical activity, and education | Plasma sCD36 is associated with T2DM risk, but not independent of adiposity. |
| Jiang et al. (2017) [85] | Cross-sectional study; T2DM subjects (n=357) | Age, sex, education, duration of T2DM, and hypertension | Plasma sCD36 is associated with carotid IMT in T2DM. |
| Handberg et al. (2008) [86] | Cross-sectional study; subjects with high-grade internal carotid stenosis (n=62) | NA | Plasma sCD36 is increased in patients with symptomatic or instable carotid plaques. |
| Wang et al. (2018) [87] | Case-cohort study; incident CHD cases (n=1,963) and non-cases (n=1,759); from baseline (1993–1997) to 2008 | Age, sex, BMI, smoking, alcohol, physical activity, education, self-reported hypercholesterolemia, and diabetes. | Plasma sCD36 is not associated with CHD risk in the total population. |
f/u, follow-up; sCD36, soluble cluster determinant 36; T2DM, type 2 diabetes mellitus; NA, not applicable; PCOS, polycystic ovary syndrome; BMI, body mass index; HDL-C, high-density lipoprotein cholesterol; hs-CRP, high sensitivity C-reactive protein; T1DM, type 1 diabetes mellitus; IMT, intima-media thickness; CHD, coronary heart disease.
![]()
In addition, sCD36 was associated with the prevalence of T2DM. We found that sCD36 level was consistently increased with abnormal glucose tolerance (prediabetes and T2DM) compared to normal glucose tolerance, and have suggested a novel index based on sCD36 (sCD36 index=ln [sCD36 (pg/mL)×FPG (mg/dL)/2]). sCD36 index was positively associated with the risk of T2DM even after adjusted by several risk factors [77], so it would help to distinguish the risk of diabetes.
Although the exact nature of plasma sCD36 is still unclear, both non-cell bound form and tissue expression are known to be associated with metabolic disorders. Regarding the nature of sCD36, recent studies showed that it is associated with circulating microvesicles [78], which are a subset of extracellular vesicles. Bariatric surgery significantly reduced the hepatic CD36 expression [79] as well as the levels of CD36-bearing microvesicles of monocyte and endothelial origin accompanying improvements in metabolic phenotypes and low-grade inflammation following weight loss [80]. In peripheral blood mononuclear cells, the CD36 expression was increased in patients with T2DM, but not in obese subjects, and metformin treatment was associated with low CD36 levels [81]. It is inferred the relationship between sCD36 and diabetic vascular complications from previous experimental studies, one small group study demonstrated that both plasma and urine sCD36 levels were increased in diabetic patients with albuminuria and it was suggested as an adjunctive marker for diabetic nephropathy [82].
However, some have raised questions about the direct link between sCD36 as a biomarker of cardio-metabolic disorders. Recent studies suggested that sCD36 had a weak correlation with T2DM and was not associated with type 1 diabetes mellitus [83], and sCD36 level was associated with risk of incident T2DM, but was not independent of BMI, waist circumference, or body fat percentage [84]. They suggest that sCD36 may be associated with insulin resistance rather than T2DM itself.
Regarding atherosclerotic cardiovascular disease, according to two cross-sectional studies, carotid intima-media thickness (IMT) showed a J-shape relation according to sCD36 quartiles in non-diabetic healthy participants [73] and sCD36 significantly increased the risk of increasing carotid IMT after multivariate adjustments among T2DM patients [85]. In a small group study of patients with high-grade internal carotid stenosis, the group with clinical symptom within the last two months had significantly higher sCD36 levels than the group with symptoms between 2 and 6 months or asymptomatic, but HbA1c levels didn't differ among groups [86]. However a large prospective case-cohort study demonstrated that elevated plasma CD36 levels were not associated with higher coronary heart disease (CHD) risk after adjusting risk factors and showed a suggestive positive association with higher CHD risk only in participants with T2DM [87].
Studies on the association of CD36 genotype with T2DM and complications are also underway. CD36 rs3211938 (T/G) polymorphism was associated with T2DM in North Indian population [88], and CD36 rs1761667 and rs10499859 polymorphisms were associated with ischemic stroke after adjustment of metabolic factors in Chinese Han [89]. There is also an opinion that the CD36 genotype is associated with obesity and thus indirectly affects progression to T2DM [90].
Taken together, CD36 is associated with cardiometabolic disorders such as metabolic syndrome, T2DM, and its complications. Plasma CD36 would be a surrogate marker for screening peoples at risk, but more large-scale studies are warranted to validate whether it is directly or indirectly linked.
Go to :
CONCLUSIONS
Experimental evidence suggests that CD36 contributes to pancreatic β-cell dysfunction and damage caused by glucolipotoxicity in diabetes. Clinical studies also supported these shreds of evidence that CD36 is closely linked with metabolic disorders. However, the mechanisms underlying β-cell dysfunction and death in diabetes are complex. Understanding this complex scenario and the role of CD36 activated redox-regulated pathways could have considerable impact on the treatment of β-cell failure and T2DM and answers to several key questions remain elusive. Thus, additional studies generate animals with a genetically engineered CD36 expression in pancreatic β-cells are required to determine the link between CD36 and β-cell failure. This may potentially lead to the development of novel therapeutics to prevent and treat T2DM.
Go to :
 XML Download
XML Download