

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Histone deacetylases (HDACs) are the enzymes that catalyze the removal of acetyl groups from ε-N-acetyl-lysine on histone proteins H2A/B, H3, and H4. When acetyl groups are removed from histones, DNA molecules wrap up histone proteins more tightly, consequently inhibiting gene transcription [1]. Conventionally, histone proteins are known as unique HDAC substrates. However, studies have identified non-histone substrates of HDAC, which are involved in the regulation of cell cycle progression, cytoskeleton reorganization, RNA splicing, and nuclear transport [2].
HDAC are categorized into five major classes based on their sequence homology to the yeast original enzymes [3]. Class I HDAC include HDAC1, HDAC2, HDAC3, and HDAC8; Class IIa HDAC are HDAC4, HDAC5, HDAC7, and HDAC9; and Class IIb HDAC are HDAC6 and HDAC10. Class III HDAC are also known as NAD+-dependent sirtuin 1–7. HDAC11 is the only class IV HDAC. Similar to other class IIa HDAC, HDAC9 shuttles in and out of the nucleus, which is a significant way to regulate its activity [4]. Also, while Class I, III, and IV HDAC are ubiquitously expressed, HDAC9 is abundant in the brain and skeletal muscles with lower expression in the liver, bone marrow, and spleen in humans [56].
Six different transcriptional variants of human HDAC9 have been identified to date [567]. The full-length form of HDAC9 is composed of 23 coding exons, which can be translated into a polypeptide containing 1,011 amino acids [5]. Zhou et al. [7] reported an uncharacterized human protein that shares a 50% identity with the N-terminal amino acid sequence of HDAC4, which was designated as HDAC-related protein (HDRP). HDRP, a truncated splice isoform of HDAC9 that lacks of the acetylase domain, is also called myocyte enhancer factor-2 (MEF2)-interacting transcription repressor (MITR). HDRP is recognized as a transcriptional repressor that shares 50% amino acid sequence identity to human HDAC4 and HDAC5 and a homolog of Xenopus MITR [7]. A year later, the same research group reported the sequence of full-length HDAC9 along with another alternative splicing product of HDAC9, i.e., HDAC9a [5]. Compared to the full-length form, HDAC9a is 132 amino acids shorter at the C terminus. Petrie et al. [6] cloned three more splicing variants of HDAC9 , i.e., HDAC9Δexon7, HDAC9Δexon12, and HDAC9Δexon15, in COS cells, a monkey fibroblast-like cell line.
Phenotypic analyses in global Hdac9 knockout and Hdac9 transgenic mice suggest that HDAC9 is a regulator of myocyte [8] and adipocyte differentiation [9] and it plays a crucial role in the development of cardiac muscle [10]. Moreover, HDAC9 has been linked to the pathological processes of human diseases, such as cardiovascular disease (CVD), autoimmune disease, and breast cancer [11121314]. The present review summarizes the current understanding of the regulation and functions of HDAC9, and its roles in the development of chronic diseases.
Go to : 
ROLE OF HDAC9 IN GENE TRANSCRIPTION
Based on the role of HDAC in chromatin remodeling, HDAC9 is generally considered as a negative regulator of gene transcription. It was shown that the transcriptional activity of Gal4 reduces when Gal4 co-expresses with HDRP in HEK293T cells [7]. This result indicates that HDAC9 can suppress gene transcription. Interestingly, genome-wide mapping data suggest that HDAC are involved in gene activation in human breast epithelial cell lines and primary T-cells [1516]. It is possible that HDAC9 also contributes to transcription activation.
Repressive effect of HDAC9 on transcription by the induction of histone deacetylation
HDAC9 may induce histone deacetylation to inhibit gene transcription by functioning as a scaffold in a transcriptional repressor complex [17]. It is commonly believed that HDAC induce histone deacetylation in the promoter of target genes to suppress gene transcription, when they are recruited by transcription factors or incorporated into multiprotein transcriptional complexes. Class IIa HDAC contain a predicted deacetylase activity in their C-terminus [18]. However, in vitro assays suggest that HDAC4, 5, and 7 lack intrinsic enzymatic activities [1920]. The HDAC activity of human HDAC9 isolated from HEK293T cells was approximately 10% of human HDAC4 enzyme activity, indicating that HDAC9 has low intrinsic HDAC activity [5]. Class I, IIb and IV HDAC possess a conservative catalytic tyrosine residue in their catalytic domain [21]. However, this tyrosine residue is replaced by histidine in vertebrate class IIa HDAC, which results in the reduction of their deacetylase activity [21]. It has been shown that the enzyme activity of class IIa HDAC relies on the association of their C-terminus with a transcriptional repressor complex containing HDAC3, silencing mediator for retinoid and thyroid receptors/nuclear receptor corepressor, and nuclear receptor co-repressor [1920]. Therefore, HDAC9 may be a pseudoenzyme and function as a scaffold for the recruitment of HDAC3 to a transcriptional repressor complex as do other class IIa HDAC [1922].
All members of class II HDAC, including HDAC9, contain binding sites for MEF2 family in the N-terminus [23]. MEF2 family is transcriptional factors essential for myogenic differentiation [24]. According to Mejat et al. [25], MITR physically interacts with HDAC1 and HDAC3, attenuating denervation-induced histone H3 hyperacetylation and reduces the transcriptional activity of MEF2 in murine skeletal muscle. Skeletal muscle from Hdac9-deficient mice showed increased histone H3 acetylation on the promotor of MEF2 target genes compared to that of wild-type (WT) mice [25]. Also, overexpression of WT Mitr reduced MEF2 transcriptional activity while overexpression of mutant Mitr lacking in the MEF2 interaction domain abolished the reduction in mouse skeletal muscle [25]. This study demonstrates that HDAC9 is likely to suppress MEF2 transcriptional activity by inducing histone H3 deacetylation on the promoter of its target genes via its interaction with MEF2.
C-terminal-binding protein (CtBP), a widely-expressed transcriptional co-repressor, is shown to be involved in the repressive effect of HDAC9 on the transcriptional activity of MEF2. MITR physically interacts with CtBP in a cell-free system as well as in HEK293T cells [26]. Compared to WT Mitr, transfection of mutant Mitr without CtBP binding activity had less suppression on MEF2C transcriptional activity by approximately 50% in 10T1/2 cells, a mouse fibroblast cell line, indicating that its binding to CtBP is required to inhibit the transcriptional activity of MEF2 [26]. Interestingly, the absence of CtBP binding activity did not inhibit MITR binding to other HDAC, including HDAC1, HDAC3, HDAC4, and HDAC5, in HEK293T cells. Therefore, MITR may repress MEF2 transcriptional activity by binding to other class I and IIa HDAC independent of CtBP.
Deacetylation of transcription factors by HDAC9 for the suppression of the transcriptional activity
Studies have shown that HDAC9 deacetylates transcriptional factors, resulting in the reduction of their transcriptional activity. Wong et al. [27] identified upstream stimulatory factor 1 (USF1), a transcription factor, as the first non-histone substrate of HDAC9. USF1 is essential for the transcriptional activation of lipogenic genes, such as fatty acid synthase (FASN) and mitochondrial glycerol-3-phosphate acyltransferase (GPAM), as sterol regulatory element-binding proteins-1c cannot bind its response elements without being recruited by USF1 [28]. In the absence of insulin, HDAC9 directly binds to USF1, which is constitutively bound to the promoter region of FASN and GPAM in cell-free conditions, HEK293F cells, HepG2 cells (a human hepatocellular carcinoma [HCC] cell line), and fasted mouse livers [27]. Transfection of HDAC9 in HEK293F cells inhibited acetylation in Lys237 of USF1, reducing the transcriptional activity of USF1. Signal transducer and activator of transcription 5 (STAT5), a transcription factor mediating interleukin 2 signaling [29], is another non-histone HDAC9 substrate. The acetylation and transcriptional activity of STAT5 increased in regulatory T-cells (Treg) from Hdac9 knockout mice when comparing to those from WT mice, indicating that HDAC9 deacetylates STAT5 to inhibiting the formation of transcriptionally active STAT5 dimer [30].
Deacetylation of transcriptional factors by HDAC9 may also facilitate their degradation. In addition to acetylation, a lysine ε-amino group is also a site for other post-translational modifications, such as ubiquitination and sumoylation, which can facilitate protein degradation [31]. Acetylation in the lysine ε-amino group can stabilize proteins by competing with ubiquitination, consequently inhibiting proteasome-mediated protein degradation [32]. NK3 homeobox 2 (Nkx3.2) is a transcription factor that inhibits chondrocyte hypertrophic maturation during cartilage development [33]. Choi et al. [34] suggest that HDAC9-mediated deacetylation induced sumoylation and subsequent ubiquitination of Nkx3.2, resulting in its degradation in ATDC5 cells, a mouse chondrocyte cell line. It is likely that HDAC9 functions as a scaffold to recruit HDAC1 to deacetylate Nkx3.2 although it physically associates with Nkx3.2.
Potential role of HDAC9 in transcriptional activation
While HDAC are commonly regarded as suppressors of gene transcription, a previous genome-wide mapping study suggests that HDAC can also play a role in transcriptional activation [16]. HDAC1, 2, 3, and 6 are enriched in the chromatin of active genes, but not silent genes, with positive correlation with gene transcription in human T helper cells [16]. As histone hyperacetylation in transcribed regions can lead to chromatin destabilization and cryptic initiation of transcription, HDAC are required to keep an appropriate level of histone acetylation to poise genes for future activation [16]. Also, some transcription factors, such as β-catenin and CCAAT/enhancer-binding protein β, are activated by deacetylation [3536]. Our RNA-Seq analysis showed that Hdac9 deletion in primary mouse hepatic stellate cells (HSC) suppressed several genes while upregulating considerable numbers of genes (unpublished data). One possible explanation is that HDAC9 may inhibit the expression of transcriptional repressors.
Go to :
MECHANISMS FOR THE REGULATION OF HDAC9 ACTIVITY
The activity of HDAC9 is tightly regulated transcriptionally, post-transcriptionally, and post-translationally as described below.
Transcriptional regulation of HDAC9 expression
While transcriptional regulation is an essential means to alter the activity of HDAC9, limited studies have been conducted to determine transcription factors that can control the transcription of HDAC9. During cardiac and skeletal muscle differentiation, Hdac9 transcription is up-regulated when myoblasts enter their differentiation pathway [37]. Members of the MEF2 family, i.e., MEF2A, MEF2C, and MEF2D, bind to the promoter of Hdac9 for the induction of its transcription [37]. Interestingly, although the MEF2 family is well-known transcription factors of all class IIa HDAC, they bind to different regions of class IIa HDAC genes. For instance, the MEF2 family bind to the enhancer regions of HDAC4 and HDAC7 while they bind to the promoter-proximal region of HDAC5 and HDAC9 [38]. Other transcription factors, including nuclear factor kappa-light-chain-enhancer of activated B-cells, early growth response protein 1, and paired-box containing 5, are suggested to induce HDAC9 expression [38]. However, future studies should be warranted to verify the role of the transcription factors mentioned above in the regulation of HDAC9 expression.
Post-transcriptional regulation of HDAC9 expression
In contrast to full-length HDAC9, its splicing variants do not contain post-translational modification sites, nuclear localization signal (NLS), or catalytic domain [567]. For instance, HDAC9Δexon7 lacks two serine residues in the NLS domain, resulting in its constitutive nuclear exclusion [6]. The removal of exon12 during splicing leads to the formation of HDAC9Δexon12, which cannot be sumoylated due to the lack of sumoylation sites. HDAC9 with partial deletion of exon 15, a possible product of RNA editing, loses a region within the deacetylase domain and the open reading frame of HDAC9 [6]. More studies are needed to determine the biological consequence of each splicing variants that may not be properly regulated at the post-transcriptional level.
Regulation of HDAC9 activity by phosphorylation
As a class IIa HDAC, HDAC9 generally exerts its function in the nucleus, and therefore its cytoplasmic retention may inhibit its activity [39]. Phosphorylation of HDAC9 is known to modulate its activity by altering the subcellular localization of HDAC9. For entry into the nucleus, the NLS of class IIa HDAC needs to bind to importin-α [38], which is an adaptor protein responsible for the nuclear import of proteins [40]. The phosphorylation of serine residues in the NLS sequence reduces its binding to importin-α, and thus inhibits the nuclear localization of class IIa HDAC [38]. Deng et al. [41] reported that minibrain-related kinase phosphorylates the NLS sequence in MITR at Ser243, leading to its cytoplasmic retention and, ultimately, the attenuation of MEF2 transcriptional suppression in C2C12 myoblasts. Ser253 in the NLS sequence of MITR has been identified as a substrate for protein kinase C-related kinase-1 [42]. While phosphorylation at this serine residue alone did not lead to MITR nuclear export, it was a priming step for the nuclear exclusion of MITR by calcium calmodulin-dependent protein kinase (CaMK) in COS cells [42].
Moreover, phosphorylation of HDAC9 can facilitate its physical interaction with 14-3-3 chaperones [43]. Binding with 14-3-3 chaperones can mask the NLS, thereby preventing the nuclear translocation of HDAC9 [44]. In COS cells, CaMK phosphorylates MITR at Ser218 and Ser448, which facilitates 14-3-3 binding to disrupt the physical interaction between MITR and MEF2 [39]. Nevertheless, the transfection of constitutively activated CaMK changed the nuclear distribution of MITR, rather than inducing nuclear exclusion. These findings support that both phosphorylation in the NLS and 14-3-3 binding are required for the nuclear exclusion of MITR.
Go to :
HDAC9 AND CHRONIC DISEASES
Zhang et al. [45] developed Hdac9 global knockout mice in 2002, which profoundly contributed to our understanding of HDAC9 functions in vivo. Mice with global Hdac9 deficiency showed no sign of cardiac abnormalities during early ages, but they developed spontaneous cardiac hypertrophy approximately 8 months after birth, indicating that the animals were sensitive to age-related hypertrophic signals [45]. Recent studies have also suggested the roles of HDAC9 in the development of various diseases, such as CVD, osteoporosis, autoimmune disease, cancer, obesity, and liver fibrosis, as described below.
CVD
Single nucleotide polymorphism (SNP) of HDAC9 is related to CVD risk in humans. Genome-wide association study (GWAS) for ischemic stroke in Europeans found that a SNP in HDAC9 (rs11984041) is positively associated with the risk of large vessel stroke [46]. Although later GWAS in the Chinese Han population did not find this SNP, another SNP of HDAC9, rs2389995, was found to be associated with decreased risk of ischemic stroke while rs2240419 was linked to the increased risk of ischemic stroke [47]. Also, compared with apolipoprotein E (ApoE) knockout mice, Hdac9 and ApoE double knockout mice exhibited remarkable decreases in aortic lesion sizes with less advanced lesions [48].
Recent studies have been conducted to elucidate the underlying mechanisms by which HDAC9 contributes to increased susceptibility and progression of CVD. Cao et al. [49] demonstrated that Hdac9 deletion resulted in the attenuation of foam cell formation and atherosclerosis progression in low-density lipoprotein receptor (Ldlr) knockout mice. The atheroprotective effect of Hdac9 deletion occurred concomitantly with increased histone acetylation at the promoters of ATP-binding cassette transporter A1 (Abca1) and ATP binding cassette subfamily G member 1 (Abcg1) in thioglycollate-elicited mouse peritoneal macrophages, resulting in enhanced macrophage cholesterol efflux [49]. Furthermore, Han et al. [11] reported that HDAC9 knockdown inhibited oxidized-LDL-induced endothelial cell apoptosis in human umbilical vein endothelial cells (HUVECs), possibly decreasing the expression of pro-inflammatory cytokines, such as tumor necrosis factor-α and monocyte chemoattractant protein 1. Therefore, data available to date indicate that inhibition of HDAC9 may be a target to decrease CVD risk.
Osteoporosis
Evidence exists to support that HDAC9 inhibits osteoclast differentiation, which may inhibit bone resorption and thus protect against osteoporosis. Jin et al. [12] reported that Hdac9 knockout mice exhibited a lower bone mass and increased serum bone resorption markers compared to WT counterparts. Also, in vitro primary mouse osteoclast differentiation was facilitated by Hdac9 deletion, but diminished by Hdac9 overexpression [12]. This HDAC9 effect was linked to the transcriptional inhibition of peroxisome proliferator-activated receptor-gamma (Pparg), which is a critical regulator of osteoclast differentiation [1250].
HDAC9 may protect against the development of age-related osteoporosis by promoting osteoblastogenesis of mesenchymal stem cells (MSC). According to Li et al. [51], a computational analysis predicts that microRNA-188 (miR-188) directly targets Hdac9 mRNA [49]. Overexpression of miR-188 suppresses Hdac9 translation in primary mouse bone marrow MSC (BMSC). In contrast, miR-188 deletion in mice reduced age-associated bone loss, higher osteoblastic bone formation, and lower bone marrow fat accumulation. Moreover, overexpression of miR-188 promoted differentiation of primary mouse BMSC into adipocytes (i.e., adipogenic differentiation) while inhibiting their differentiation into osteoblasts (i.e., osteogenic differentiation) [51]. The findings indicate that miR-188 may inhibit osteogenesis by silencing Hdac9. Also, Chen et al. [52] reported that overexpression of MITR favors osteogenesis but inhibits adipogenesis, while MITR knockdown leads to the opposing effects in 3A6 cells, a human MSC cell line, and primary human BMSC. Compared to being transfected alone, PPARγ transcriptional activity was reduced when it was co-transfected with MITR in 3A6 and HEK293 cells [52]. PPARγ facilitates MSC adipogenic differentiation while inhibits osteogenic differentiation; therefore MITR may promote MSC osteogenic differentiation by repressing PPARγ transcriptional activity [5253]. Hdac9 deficient mice appeared to have a decreased serum osteoblast number compared to their WT littermates, further supporting that HDAC9 facilitates osteoblastogenesis [12]. Overall, the evidence described above indicates that HDAC9 prevents osteoporosis by promoting osteogenesis while inhibiting osteoclastogenesis. However, data from human samples or clinical trials are needed to confirm these findings in humans.
Autoimmune disease
Previous reports suggest that HDAC9 is related to autoimmune diseases both in mice and humans. HDAC9 transcription was significantly increased in blood CD4+ T-cells from lupus patients compared with healthy controls [13]. Also, Hdac9 transcription was increased with lupus progression in total splenocytes and splenic CD4+ T-cell population from a murine disease model [13]. Furthermore, compared to control mice, colitis induced by dextran sodium sulfate was attenuated in mice with Hdac9 deficiency [54].
The contribution of HDAC9 to autoimmune diseases was linked to its inhibitory effect on the immunosuppressive function of Treg. Forkhead box P3 (Foxp3) is a key regulator of the development and function of Treg [55]. Hdac9 deficiency in mice resulted in increased Foxp3+ expression in the colon and elevated the immunosuppressive function of Treg in vitro, consequently ameliorating colitis development [56]. Moreover, Hdac9 deficiency increased Treg proliferation and function by enhancing mitochondrial energy metabolism in mice via increased MEF2 binding to the promoter of PPARγ coactivator 1-alpha (PGC-1α), a principal regulator for mitochondrial biogenesis, with a concomitant increase in sirtuin 3 (SIRT3) protein [57]. PGC-1α and SIRT3 then increased oxidative phosphorylation in mouse Treg, which is essential for the proliferation and immunosuppressive function of Treg.
Cancer
Potential roles of HDAC9 in cancer progression have emerged recently. Huang et al. [14] demonstrated that Chinese female patients with high HDAC9 expression in their breast cancer tissues had reduced overall survival compared to those with a low HDAC9 level. In addition, HDAC9 expression was significantly upregulated in the breast cancer tissues collected from patients and human breast cancer cell lines (MCF-7, BT474). Importantly, HDAC9 knockdown using small interfering RNA (siRNA) significantly suppressed cell proliferation, migration, and invasion of MCF-7 and BT474 cells [14]. HDAC9 may promote breast cancer aggressiveness by suppressing the expression of miRNA that may contribute to the development of breast cancer. Breast cancer tissues with triple-negative breast cancer (TNBC), one of the most aggressive subtypes of breast cancer, have higher HDAC9 protein levels compared to non-TNBC cancer tissues [58]. Pan-class IIa HDAC inhibitor treatment or HDAC9 knockdown reduced the invasiveness while increasing miR-206 expression in human TNBC cell lines, e.g., MDA-MB-231, and HCC1143, and inhibited the angiogenesis of tumors in nude mice [58]. As miR-206 has been identified as a critical suppressor of TNBC [59], this study suggests that HDAC9 suppresses miR-206 expression in breast cancer cells and thereby promotes breast cancer cell invasion and breast cancer tissue angiogenesis.
In addition to breast cancer cells, HDAC9 is known to suppress miRNA expression in other cancer cells. For instance, miR-376a was found to be down-regulated in HCC specimens [60]. Moreover, overexpression of miR-376a inhibited proliferation while inducing apoptosis in Huh7 cells, a human hepatocarcinoma cell line, indicating that miR-376a may inhibit the development of HCC [60]. HDAC9 knockdown in Huh7 cells remarkably increased miR-376a expression, suggesting that HDAC9 may repress the expression of miR-376a [61]. The knockdown also increased H3 histone acetylation at Lys18 in the differentially methylated region of maternally expressed 3 (Meg3-DMR), which is located upstream of the miR-376a gene, indicating HDAC9 may inhibit miR-376a expression by deacetylating Meg3-DMR [61].
Obesity and insulin resistance
Recent studies have established a link between HDAC9 and obesity/insulin resistance. Chatterjee et al. [9] demonstrated that HDAC9 functions as a negative regulator of adipogenic differentiation and Hdac9 expression is reduced in human and mouse pre-adipocytes before their adipogenic differentiation at both mRNA and protein levels. However, the reduction in Hdac9 expression before adipogenic differentiation was impaired in pre-adipocytes from high-fat diet-fed mice [62]. HDAC9 may mediate high-fat diet-induced inhibition of adipogenic differentiation. Hdac9 deletion protected mice from high-fat diet-induced weight gain, adipose tissue dysfunction, and liver steatosis while improving insulin sensitivity [62].
Obesity-induced insulin resistance is known to lead to increases in gluconeogenesis via the activation of forkhead box O1 (FOXO1) [63], which is a transcription factor phosphoenolpyruvate carboxykinase (PEPCK) and glucose 6-phosphatase (G6PC) [64]. In chronic hepatitis C virus (HCV)-infected transgenic mice and patients, hepatic HDAC9 mRNA and protein levels were increased, and the patients had increased transcription and enzymatic activity of hepatic PEPCK [65]. HCV infection up-regulates the transcription of gluconeogenic enzymes, i.e., PEPCK and glucose G6PC, while HDAC9 knockdown diminished the induction in Huh7 cells [66]. Therefore, it seems that HDAC9 may mediate the HCV infection-induced gluconeogenic gene expression. Overexpression of HDAC9 leads to FOXO1 deacetylation and increases its transcriptional activity in Huh7 cells, while HDAC9 knockdown does the opposite [66]. These results suggest that HDAC9 increases FOXO1 transcriptional activity, possibly by deacetylation for the induction of gluconeogenic gene expression.
Insulin treatment and refeeding increased the cytoplasmic retention of HDAC4 in primary mouse hepatocytes and HDAC4 phosphorylation in mouse livers [67]. Insulin signaling can activate salt-inducible kinase 2, which phosphorylates all class IIa HDAC for their cytoplasmic retention [68]. Therefore, it is possible that HDAC9 contributes to FOXO1-dependent upregulation of gluconeogenesis when insulin resistance occurs. Nevertheless, further experiments are required to test this hypothesis in animal models with diet-induced obesity and insulin resistance and human subjects with type 2 diabetes mellitus.
Liver fibrosis
HDAC9 may play essential roles in the activation of HSC and liver fibrosis. Activated HSC are major scar tissue-producing cells upon hepatic injury [69]. Our laboratory reported that HDAC9 mRNA and protein levels in primary mouse HSC increased during their in vitro activation [70]. Also, we demonstrated that HDAC9 mRNA and protein were up-regulated in human livers with primary biliary cirrhosis, nonalcoholic steatohepatitis, and alcoholic cirrhosis when compared to healthy livers [70]. Moreover, HDAC9 knockdown in LX-2 cells, a human HSC cell line, significantly decreased the expression of fibrogenic genes, such as alpha-actin 2 and collagen type I alpha one chain upon the stimulation by transforming growth factor β1, a potent fibrogenic cytokine. The studies further support that HDAC9 may be critical for HSC activation, and thus, inhibition of HDAC9 in HSC may prevent the development of liver fibrosis. An investigation is currently underway to test this hypothesis in our laboratory.
Go to :
CONCLUSIONS
By catalyzing the deacetylation of histones and transcription factors, HDAC9 plays a critical role in the regulation of gene expression (Fig. 1). While it is prevalent that HDAC9 silences gene expression, studies also exist to support that HDAC9 may be involved in transcriptional activation, although the underlying mechanism has been elusive. Mounting evidence suggests that HDAC9 is involved in the pathogenesis of CVD, osteoporosis, autoimmune disease, cancer, obesity, and liver fibrosis by altering the expression of genes critically involved in the pathogenic processes (Fig. 2). Therefore, HDAC9 may be a great molecular target for the prevention and therapy for the diseases. However, there are some challenges to overcome. Human HDAC9 gene encodes multiple protein isoforms and the role of each HDAC9 isoform in the development of chronic diseases has not been clearly understood. Also, the molecular mechanisms by which HDAC9 regulates the expression of mRNA and miRNA need further investigation.
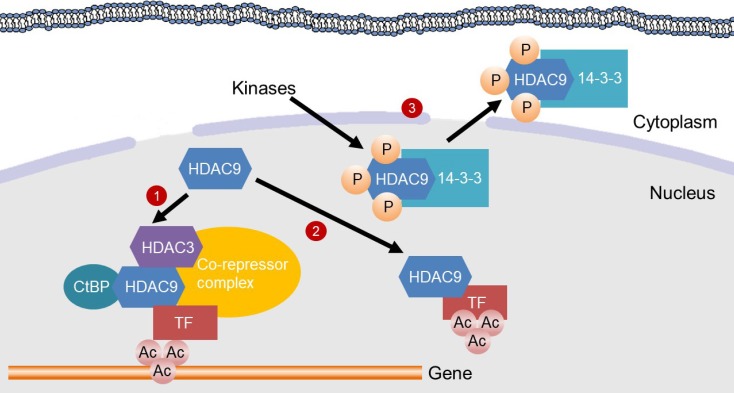 | Fig. 1Mechanisms for the inhibition of gene transcription by histone deacetylase 9 (HDAC9). HDAC9 inhibits gene transcription by inducing deacetylation of transcription factors (TFs) or in the promoter region of genes. (1) HDAC9 directly binds to C-terminal-binding protein (CtBP) and HDAC3, which are recruited to a TF to assemble a co-repressor complex. Then, the co-repressor complex deacetylates histones in the promoter region of a gene, thereby suppressing gene transcription. (2) HDAC9 binds to a TF for its deacetylation, which inhibits the transcriptional activity. (3) Kinases, such as minibrain-related kinase and calcium calmodulin-dependent protein kinase, phosphorylate the nuclear localization signal region of HDAC9 for subsequent 14-3-3 binding, which leads to the exit of HDAC9 from the nucleus and therefore a loss of its activity.
|
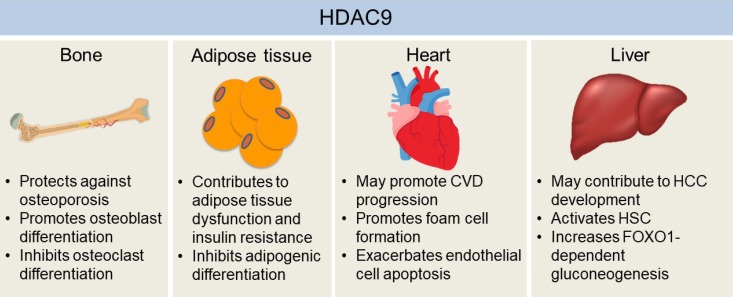 | Fig. 2The effects of histone deacetylase 9 (HDAC9) on the development of chronic diseases in bone, adipose tissue, heart, and liver. HDAC9 facilitates osteoblast activity while inhibiting osteoclast differentiation, which may explain the protective effects of HDAC9 against osteoporosis in mice. Adipogenic differentiation of pre-adipocytes may help to alleviate hypoxia and dysfunctions of hypertrophied adipose tissue, which is inhibited by HDAC9. Also, mice with the deletion of Hdac9 are protected from diet-induced adipose tissue dysfunctions and systemic insulin resistance. Hdac9 deficiency also enhances cholesterol efflux from macrophages while inhibiting oxidized low-density lipoprotein-induced human endothelial cell apoptosis, indicating HDAC9 may exacerbate cardiovascular disease (CVD) development. Moreover, HDAC9 may contribute to hepatocellular carcinoma (HCC) development by suppressing miR-376a expression, a microRNA that induces the apoptosis of hepatocellular carcinoma cells. HDAC9 is also critical for hepatic stellate cell (HSC) activation and forkhead box O 1 (FOXO1)-dependent gluconeogenesis in the liver. Therefore, it may also be involved in the development of fibrosis and obesity-induced insulin resistance in the liver.
|
Go to :
 XML Download
XML Download