

 Citation
Citation Print
Print



Autosomal recessive limb-girdle muscular dystrophy (LGMD), which constitutes a group of clinically and genetically heterogeneous muscular disorders, has been linked to 26 genes. Type-2C LGMD (LGMD2C) to LGMD2F are due to variants in the diverse genes that encode proteins of the sarcoglycan complex: α-, β-, γ-, and δ-sarcoglycans. These four sarcoglycans are components of a major dystrophin-glycoprotein complex that stabilizes the plasma membrane and cytoskeleton from the mechanical stress of contractile activity and is involved in signal transduction [1]. The variants associated with the sarcoglycan gene characterize early childhood-onset progressive weakness that overlaps the spectrum associated with dystrophin variants in Duchenne and Becker muscular dystrophy with the relatively milder form LGMD [2,3]. Since LGMD subtypes are difficult to distinguish based on clinical manifestations, muscle imaging, and laboratory tests such as immunoblotting, immunohistochemical analysis is used to determine the patterns of structural changes and protein deficiency. Subsequent molecular analysis is performed to identify specific genetic defects and the sites of chromosomal lesions.
Go to : 
CASE
This study reviewed two siblings diagnosed with LGMD2E with recessive inheritance patterns (Fig. 1A). The proband was a 43-year-old female (II-8) who showed delayed early motor milestones in crawling, sitting, and walking. She was the normal product of a full-term pregnancy, but at 8 years she showed a waddling gait and was never able to run. The patient used a wheelchair in her early teens due to the progression of foot deformity and motor difficulty. Her younger sibling was a 41-year-old female (II-9) who showed a slightly milder but otherwise similar clinical prognosis. She had reached her early developmental milestones as expected, but her mother reported that she had difficulty sitting up at 4 years of age, with hypertrophy at the calves. The patient walked using a tiptoe gait as a result of shortening of the Achilles tendon, and exhibited a gradual loss of ambulation resulting in wheelchair dependency by the age of 17 years. Weakness of her upper limbs prevented her from raising her arms overhead after the third decade.
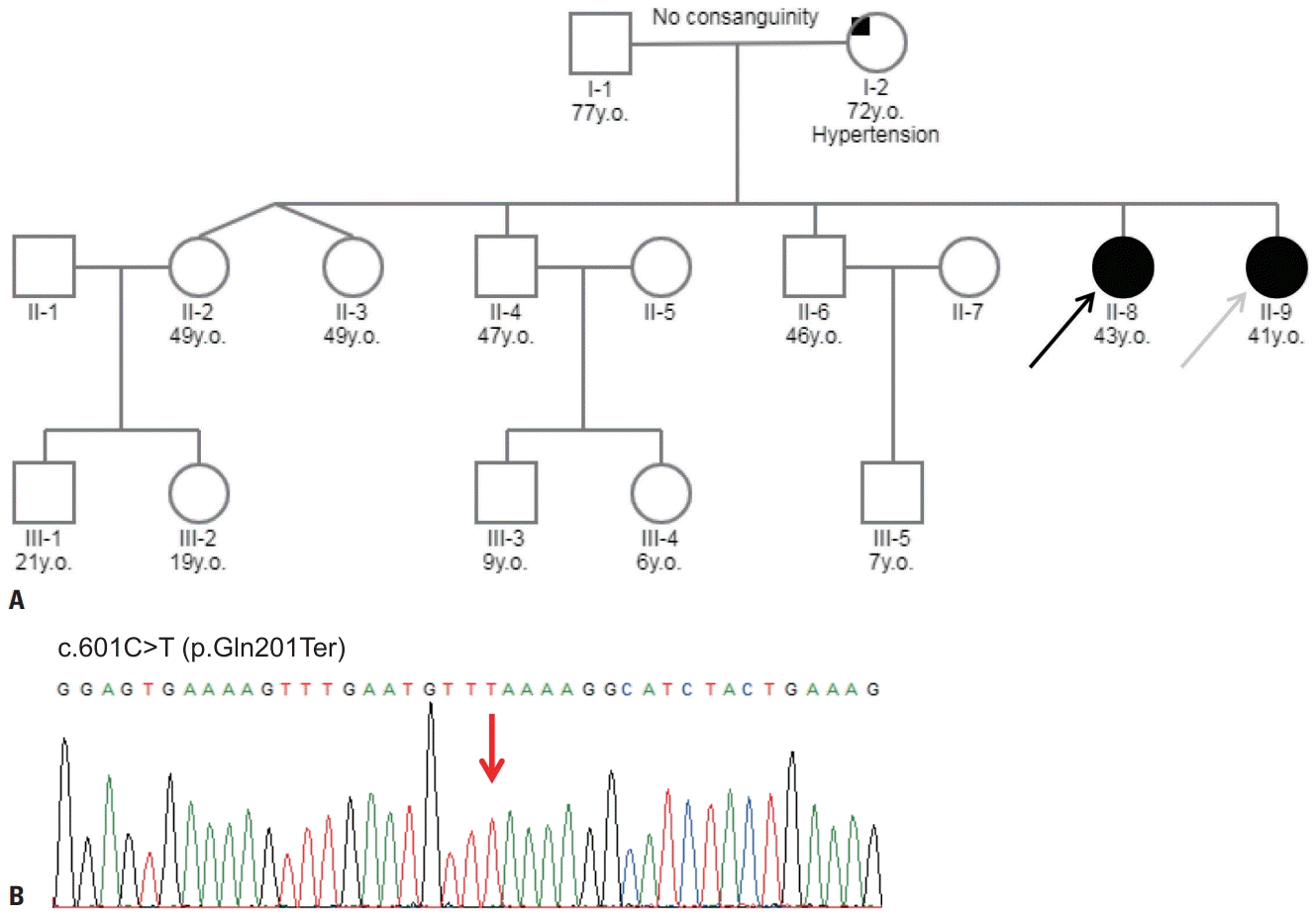 | Fig. 1.Pedigree and sequencing chromatogram of the patients with limb-girdle muscular dystrophy. (A) The family pedigree: males (squares), females (circles), affected individuals (filled symbols), unaffected individuals (open symbols). The proband (II-8, black arrow) and the younger sibling (II-9, gray arrow) had a clinical presentation of childhood muscular dystrophy. (B) Sanger sequencing chromatogram of the SGCB variant in the proband. The cDNA sequence with a homozygous C-to-T substitution in exon 4 (red arrow) was predicted to terminate transcription and produce a truncated β-sarcoglycan protein.
|
Both siblings needed noninvasive ventilator support and were confined to bed due to diffuse and symmetric muscle weakness. Physical examination revealed ankle contracture, scapular winging, and diffuse muscular atrophy with significant weakness in both proximal and distal limbs. Bulbar muscles were not involved. Their serum creatine kinase (CK) levels were 184 and 254 IU/L (normal range: 21–215 IU/L). Muscle biopsies were performed at the ages of 34 and 32 years in the proband and her sister, respectively. Pathologic studies revealed similar chronic and myopathic features, including prominent fatty replacement with a few severe atrophic or reactive hypertrophic muscle fibers. Electrocardiography and transthoracic echocardiography did not reveal any significant conduction or structural abnormalities, while electromyography findings were compatible with myopathy (Table 1).
Table 1.
Clinical and pathological features of Korean patients with nonsense variant in SGCB gene
| Patients | Sex | AAO (years) | AAE (years) | mGMW scalea | ALA (years) | FH | Calf muscle hypertrophy | Bony abnormality | Muscle pathology | Cardiologic involvement | Pulmonary function | CK |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | Neonatal | 43 | 9 | 13 | AR | (+) | (+) Winged scapula, Ankle contracture | End stage of muscle atrophy and replacement by fat tissue | (-) on EKG, TTE | FVC 12.0% | 184 |
| 2 | F | 4 | 41 | 9 | 17 | AR | (+) | (+) Winged scapular, Scoliosis, Ankle contracture | End stage of muscle atrophy and replacement by fat tissue | (-) on EKG, TTE | FVC 19.1% | 254 |
![]()
To identify the genetic cause of myopathy, we performed targeted sequencing of 212 myopathy-related genes, which identified a homozygous nonsense variant (c.601C>T;p. Gln201Ter) in SGCB (Fig. 1B). This variant is classified as likely pathogenic based on the following evidence: 1) null variant in a gene where loss of function is a known disease mechanism, 2) appearing at extremely low frequencies in controls, and 3) multiple lines of computational evidence supporting deleterious gene-induced effects. This study was approved by the local institutional review board, and written informed consent was obtained from all patients.
Go to :
DISCUSSION
Variants in sarcoglycan genes were reported recently [4], but the present report is the first to include clinical aspects of genetically confirmed cases with a likely pathogenic variant in SGCB in Korea. LGMD2E is a rare type of sarcoglycanopathies that is associated with early-onset severe muscular dystrophy, myocardial involvement, and respiratory impairment developing as the disease progresses. In the present case, the age at onset ranged from neonatal to 4 years, with the early onset of disease followed by more-rapid loss of ambulation. Both cases presented with the clinical characteristics of LGMDs: muscle weakness predominance in both axial and proximal muscles, progressing from the lower to the upper extremities and not encompassing diagnostic obstacles with other myopathies. Muscle biopsies revealed end-stage muscle atrophy, and the serum CK levels were not elevated, in proportion to the disease severity. Mechanical ventilation support was needed due to severe respiratory insufficiency following significant limb muscle weakness. All patients showed cognition preservation and no cardiac involvement even in the late stage of the disease, despite cases of cardiomyopathy in LGMD2E frequently being reported [5].
This case report has revealed a novel homozygous nonsense variant at exon 4 in SGCB (c.601C>T;p.Gln201Ter). This variant was predicted to produce a truncated β-sarcoglycan caused by the lack of an extracellular portion including all glycosylation sites, and classified as likely pathogenic according to the 2015 guidelines by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. It was not found in the ExAC or 1000G database as homozygotes, and so was anticipated to be disease-causing based on in-silico analyses implemented using VariantTaster (http://varianttaster.org). Truncating variants are known to be associated with a severe phenotype of LGMD2E [6]. Nevertheless, a missense variant in exon 3 affecting proximal extracellular domain resulting in deficiency of all three sarcoglycans (α, β, and γ) tested immunohistochemically can also cause a severe phenotype [7]. The sarcoglycan expression and clinical presentation seem to be correlated, with the absence of sarcoglycans associated with a severe phenotype such as the early loss of ambulation. Likewise, variants in any single sarcoglycan gene can lead to the absence of this protein and a secondary deficiency of all sarcoglycans. Since there is no specific immunostaining pattern for any sarcoglycanopathies [1], pathologic findings should be combined with the findings of muscle imaging, clinical presentation, and genetic analysis. Molecular genetics is a helpful tool for diagnosing muscular dystrophies, especially those with atypical clinical features.
Sarcoglycanopathies are known to be the third- to fourthmost-common LGMD subtypes, whose relative frequencies vary with ethnicity and geographical distribution. However, their prevalence has been reported to be surprisingly low in Korea [4]. LGMD2C is the most-common of the four sarcoglycanopathies in India [8], while LGMD2D is the most prevalent in Europe and North America [2,9], and LGMD2E is the second-most-common variant after LGMD2D in China [10]. Further research involving larger cohorts is needed to clarify the clinical and genetic spectrums with genotype–phenotype correlations of sarcoglycanopathies.
Go to :
 XML Download
XML Download