

 PDF
PDF Citation
Citation Print
Print



Introduction
Histiocytosis is a complex and heterogeneous group of disorders of unknown etiology. Moreover, differentiating the types of histiocytosis can be quite challenging and requires a combination of clinical, radiological, and immunohistochemical features [1]. Erdheim-Chester disease (ECD) is characterized by the accumulation of foamy macrophages, chronic inflammation, fibrosis, and organ failure [2]. ECD tumors contain lipid-laden macrophages that are CD68-positive and CD1a-/S100-negative [3]. ECD primarily affects the bones and may involve virtually every organ and tissue with protean clinical manifestations, ranging from asymptomatic bone involvement to multisystemic, life-threatening forms [2].
We describe a unique case of multi-retroperitoneal organ involvement, mainly the ureter, kidney, rectum, bladder, and uterus, with no skeletal bone involvement.
Case report
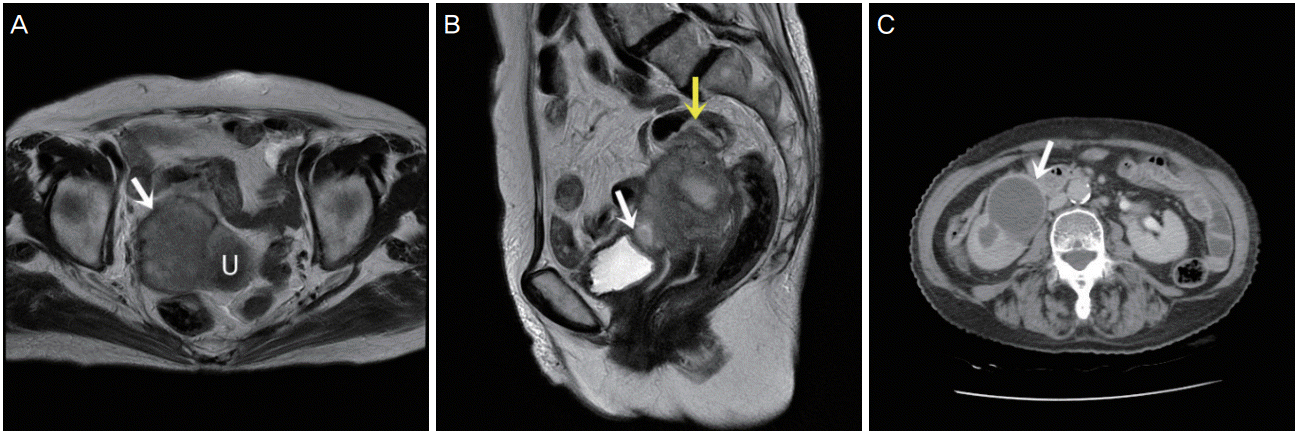
A 73-year-old woman visited our hospital with vaginal spotting for a month. She had also been experiencing abdominal pain along with pus-like urine. Transvaginal ultrasonography revealed a 5-cm mass on the anterior uterus. The mass was suspected to have invaded the bladder and rectum. Laboratory studies revealed an elevated white blood cell count of 12.5 103/μL and a C-reactive protein level of 23.17 mg/dL. However, other tumor markers (CA125, CA19-9, carcinoembryonic antigen) were within the normal range. Magnetic resonance imaging also revealed a 5.2×5.8×6.8-cm3 irregular mass lesion in the uterus that invaded the right pelvic sidewall, bladder, rectosigmoid colon, and right ureter (Fig. 1A and B).
A computed tomography urography scan of her abdomen also revealed a lobulated soft tissue mass in the uterus that invaded the right mid ureter, severe hydronephrosis, and diffuse wall thickening at the rectosigmoid junction of the colon (Fig. 1C).
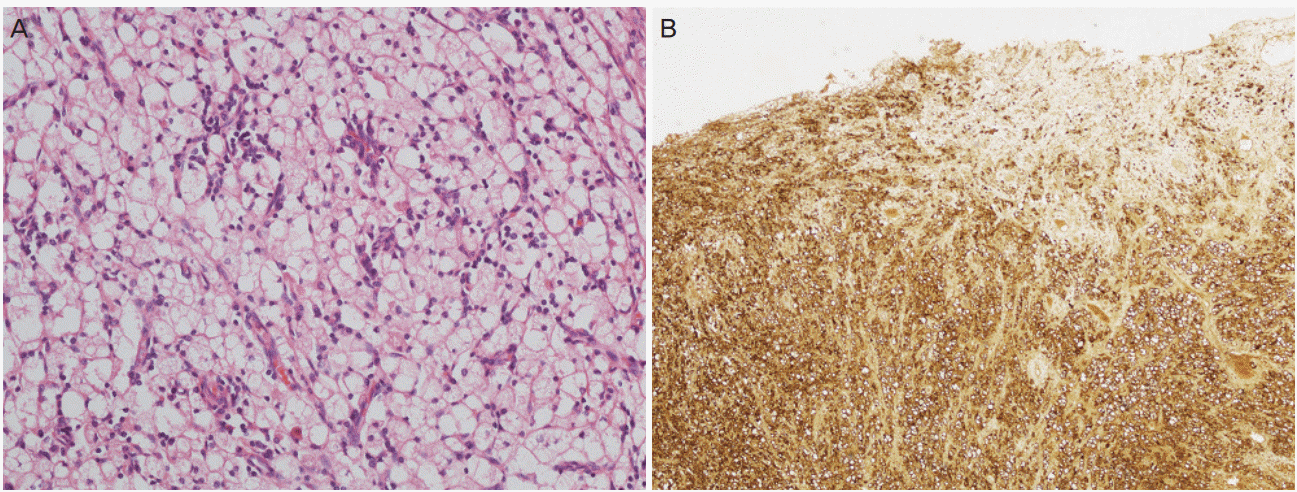
Explorative laparotomy was performed based on the suspicion of a uterine sarcoma that invaded the right ureter, rectum, and bladder. A lobulated uterine mass approximately 7 cm in size was agglomerated with the rectum and bladder and invaded the right ureter and pelvic wall. The right ureter was filled with pus-like fluid. Total abdominal hysterectomy, bilateral salpingo-oophorectomy, low anterior resection and small bowel resection, appendectomy, right nephrectomy, and partial cystectomy were performed. Frozen sections of the uterus, kidney, and rectum showed diffuse histiocytic infiltration. The final histopathology showed diffuse histiocytic infiltration within the entire myometrium, rectum, right ureter, right kidney, and bladder. Tumor cells had large amounts of cytoplasm with a foamy appearance; however, there were no atypia or mitosis (Fig. 2A). For differential diagnosis of histiocytic proliferation in the female genital tract, further diagnostic workup including immunohistochemistry (CD68, S100, and cytokeratin) and molecular pathology (BRAF V600E gene) was performed. Immunohistochemical staining was positive for CD68 (Fig. 2B) and negative for S100 and cytokeratin. BRAF V600E gene mutation was absent in realtime PCR. The bone scan revealed no uptake in long bones.
The patient was regularly followed up at 3-month intervals with no medical therapy, and she remained without evidence of recurrence 8 months after surgery.
Discussion
ECD is an extremely rare type of non-Langerhans cell histiocytosis involving multiple organs including the bones, central nervous system, heart, lung, retroperitoneum, and skin. Skeletal involvement is the most frequent manifestation, with 96% of patients displaying the characteristic radiological finding of bilateral symmetric long bone sclerosis [4]. However, ECD has heterogeneous and diverse clinical manifestations. Atypical presentations can be challenging for the diagnosis of ECD. ECD with no skeletal involvement is extremely rare, and only a few cases have been reported [5]. Our patient had no skeletal involvement.
Histologically, there is proliferation of foamy lipid-laden histiocytes surrounded by Touton giant cells. Due to the lack of cytological atypia in the cells, the neoplastic nature of the proliferation is often missed. Immunohistochemical staining for CD68, CD163, and FACTOR XIII is positive, with CD1a, Langerin, and S100 being negative. The BRAF V600E mutation is present in more than 50% of ECD cases, with other mutations such as N/KRAS and PIK3CA described in BRAFnegative ECD [6].
In a prospective investigation of 60 adults with ECD conducted at the National Institutes of Health (NIH) clinical center, one-third of patients had a history of renal impairment before their NIH visit [7]. A total of 65% of patients exhibited encasement of the kidney and involvement of the retroperitoneal space, causing renal artery stenosis in 54% of cases and ureteropelvic junction obstruction in 51% of cases [7]. However, a study investigating abdominal involvement during ECD reported that there was no uterine involvement [8].
ECD has no standard therapy, although consensus guidelines for clinical management were recently reported [9]. Empiric treatments include anti-inflammatory, immunosuppressive, and chemotherapeutic agents, and recent reports of BRAF V600E and MAPK pathway gene mutations in ECD have suggested treatment with BRAK and MEK inhibitors [7].
Our patient had multiple retroperitoneal organ involvement without skeletal bone involvement. Tissue biopsy and immunohistochemical studies suggested ECD diagnosis. However, the BRAF V600E gene mutation was not found. To the best of our knowledge, uterine involvement in ECD has not been reported. Differential diagnosis of ECD in the female genital tract includes a variety of infectious, inflammatory, and neoplastic disorders. Xanthogranulomatous inflammation involving the uterus is typically centered in the endometrium and involves a combination of neutrophils, plasma cells, lymphocytes, and hemosiderin-laden histiocytes [10]. Unlike ECD, uterine Rosai-Dorfman disease involves lymphocytophagocytosis or emperipolesis [11].
Our patient presented atypical ECD with pathologic and immunohistochemical characteristics that coincided with ECD and clinical characteristics such as skeletal bone involvement and BRAF V600E gene mutation that did not coincide with ECD. To the best of our knowledge, this is the first case report of retroperitoneal ECD with mainly uterine involvement. To correctly diagnose atypical ECD, communication among clinicians, radiologists, and pathologists is vital.
 XML Download
XML Download