

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Atherosclerosis is a complex process involving interactions among vascular cells, lipids, and inflammatory mediators [1]. A number of inflammatory mediators, including pro-inflammatory cytokines, growth factors, oxidative stress, and toxins, induce the conversion of endothelial cells (ECs) into mesenchymal fibroblast-like cells that promote disease progression, a process known as the endothelial-to-mesenchymal transition (EndMT) [2,3]. Several factors have been identified as inducers of endothelial fibrosis through EndMT, either by acting individually, via synergistic processes, or by forming an intercellular signaling pathway [4]. The most important endothelial fibrosis inducers described to date include transforming growth factor-β (TGF-β), the pro-inflammatory cytokines interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α), the transcription factor nuclear factor-kappa B, bacterial endotoxins, and oxidative stress [2–4].
IL-1β is upregulated in patients with diabetes, and endothelial dysfunction is involved in diabetic pathogenesis [2,3]. IL-1β plays a stimulatory role in the progression of atherosclerosis and has been shown to exert important pro-inflammatory and pro-apoptotic effects [3]. Among all of the known pro-inflammatory cytokines, IL-1β is the most potent inducer of EndMT [2,3]. Long-term exposure to TNF-α or IL-1β induced the permanent transformation of human dermal microvascular endothelial cells into myofibroblasts in cell culture [5]. IL-1β and TGF-β2 are also known to have a synergistic effect, and strongly promote EndMT [2,6]. Bone morphogenetic proteins (BMPs), which belong to the TGF-β superfamily, have various activities including cell proliferation, differentiation, and endocrine regulation [7]. BMP activity is critical for the differentiation of mesenchymal stem cells (MSCs) into chondrocytes or osteteoblasts [7]. Genetic studies in mice indicate that components of the BMP signaling pathway stabilize EC/vascular smooth muscle cell (VSMC) interactions and play a critical role in regulating vascular reactivity, EC function, and vascular inflammation [1,7,8]. Further insight into these mechanisms will be provided when cell-specific conditional deletion of different BMP pathway components is performed in the vasculature [9,10]. BMP can function in a proangiogenic or antiangiogenic manner, depending on its ligand concentration and cellular target, and the overall context. During vascular development, BMP signaling can also influence the differentiation of smooth muscle cells (SMCs) [2,10]. BMP2 and BMP7 inhibit the growth and migration of SMCs and promote their differentiation into a contractile phenotype [9,10].
Dipeptidyl peptidase IV (DPP-IV), a cell-surface and secreted peptidase, has been implicated in extracellular matrix (ECM) remodeling by regulating cell adhesion, migration, and angiogenesis [11]. DPP-IV inhibitors repress the proliferation of vascular SMCs and inflammatory reactions, improve endothelial function, reduce thrombogenesis [12], and have potential antiatherosclerotic properties. Linagliptin and sitagliptin are known to inhibit kidney fibrosis by suppressing EndMT [11,13]. However, linagliptin and sitagliptin have different activities, which may explain their diverse effects on EC biology [13]. DPP-IV inhibition by gemigliptin appears to be very effective against EndMT because it has a much faster association rate and slower dissociation rate than sitagliptin (fast on and fast off rate) and vildagliptin (slow on and slow off rate) [11], but its efficacy has not yet been demonstrated.
The primary aim of the present study was to assess whether the DPP-IV inhibitor gemigliptin inhibits IL-1β-mediated EndMT cascade through downregulation of the BMP/Smad signaling pathway and non-Smad signaling pathway in vascular ECs.
METHODS
Cell culture
Primary human umbilical vein endothelial cells (HUVECs, Modern Cell & Tissue Technologies, Seoul, Korea) were cultured in the EC basal medium provided in the EBM-2 Bullet Kit (Lonza, Basel, Switzerland), supplemented with 10% FBS. Cells were grown to confluence at 37°C in a humidified atmosphere of 5% CO2/95% air. Cells were used in the experiments after passage 4.
HUVECs were plated in 60-mm culture dishes at a density of 1×106 cells/dish. After 16 to 24 hours, the cells were washed with EBM-2 basal medium and antimycotics but no growth factors and then the cells were preexposed to 20 μM of gemigliptin (LG Chem Ltd., Seoul, Korea). The pretreated HUVEC cells were cultured in the EBM-2 medium containing 2% FBS, 10 ng/mL IL-1β for 24 hours.
Immunofluorescence
Cells cultured in a 4-well chamber were used for von Willebrand factor (vWF) and α-smooth muscle actin (α-SMA) immunofluorescence. Briefly, after being washed three times in Tris-buffered saline (TBS; 25 mM Tris-HCl, pH 7.6, and 150 mM NaCl), the cells were placed in methanol:acetone (1:1) for 20 minutes at −20°C. They were washed three times in TBS for 5 minutes and blocked 5% horse serum/TBS for 30 minutes at room temperature. The cells were incubated in primary antibody for overnight at 4°C and washed three times in TBS. Subsequently, the cells were incubated with the biotynated secondary antibodies for 1 hour, washed three times with TBS and mounted with mounting medium with 4′,6-diamidino-2-phenylindole (DAPI, Vector Laboratories Inc., Burlingame, CA, USA). To calculate the relative percentage of α-SMA-positive cells in total HUVECs, the numbers of total HUVECs and α-SMA-stained HUVECs were determined. The rate of vWF positivity was calculated as a percentage of total HUVECs, as described by the following equation: (number of α-SMA-positive cells/number of total HUVECs)×100. On average, 100 HUVECs were counted for each slide of experimental conditions.
Transwell assays
Cell migratory assays were performed using Transwell chambers with filter membranes of 8 μm pore size (Corning, Life Sciences, New York, NY, USA). Filter membranes were coated with 0.2% gelatin and chambers were inserted in 24-well cultured plates. HUVECs were seeded into the upper chamber (3×104 cells per well in 2% FBS in EBM-2 medium) and allowed to attach for 3 hours at 37°C. Subsequently, cells were treated with 10 ng/mL IL-1β, 10 ng/mL IL-1β plus 20 μM gemigliptin for 24 hours (medium alone, with gemigliptin were used as controls). Filter membranes were removed, fixed with 70% ethanol and stained with 1:1,000 DAPI (1 mg/mL) for 30 minutes at 37°C. Migration activity was quantified by counting the migrating cells using a florescence microscope (Carl Zeiss Co. Ltd., Oberkochen, Germany). Data are reported as the ratio of migrating versus non-migrating cells expressed as a percentage of control (untreated cells).
Wound healing assays
HUVECs seeded on 6-well plates were cultured until confluency. They were then scratched with a yellow pipette tip, washed with PBS and further incubated with 10 ng/mL IL-1β, 10 ng/mL IL-1β plus 20 μM gemigliptin in EBM-2 medium supplemented with 2% FBS (medium alone, with gemigliptin were used as controls). Five fields of each of the three wounds analyzed per condition were photographed immediately after the scratch had been made (0 hour) and 24 hours later to monitor cell movement into the wounded area.
Western blot analysis
Cells were lysed in ice-cold lysis buffer. Cell lysates were centrifuged at 15,000 rpm for 10 minutes at 4°C. Protein concentrations were measured by the BCA method using BSA as the standard. Proteins (20 μg) were separated by 8% to 10% SDS-PAGE and transferred onto nitrocellulose membranes. The membranes were blocked with 5% fat-free milk for 1 hour in TBS containing 0.1% Tween 20 (TBS-T) and then incubated with the following primary rabbit or mouse antibodies: anti-endothelial nitric oxide synthase (eNOS) and BMP receptor type II (BMP-RII, BD Bioscience, San Jose, CA, USA), vWF, and fibronectin (FN, Dako Korea LLC, Seoul, Korea), glucagon-like peptide 1 receptor (GLP-1R), transgelin (SM22), fibroblast-specific protein 1 (FSP-1), activin receptor type 1A (Act-RIA) and activin receptor type IIB (Act-RIIB), BMP receptor type IA (BMP RIA) and BMP receptor type IB (BMP-RIB), CD26 (DPP-IV), osteogenic-specific transcription factor runt-related transcription factor 2 (RUNX2), osterix, hepcidin (all from Abcam, Cambrige, UK), collagen type I (Col I) and α-SMA (Merck Millipore Co., Darmstadt, Germany), BMP2, Act-RIIA, and TGF-β receptor type II (TGFβ-RII) (all from Novous Biologicals LLC, Littleton, CO, USA), vascular endothelial cadherin (VE-cadherin), BMP4, BMP7, TGF-β receptor type I (TGFβ-RI) (all from Santa Cruz Biotechnology Inc., Dallas, TX, USA), phospho-Smad1/5/9, Smad1, phospho-Smad2, Smad2, phospho-Smad3, Smad3, phospho-extracellular regulated protein kinases (p-Erk), Erk, phospho-p38, p38, phospho-SAPK/JNK, and SAPK/JNK (all from Cell Signaling Biotechnology, Danvers, MA, USA), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH, Abclone, Seoul, Korea). The antibodies were diluted 1:500 to 1:2,000 in 1% BSA in TBS-T and incubated at 4°C overnight. After washing, the membranes were incubated with secondary peroxidase-conjugated anti-mouse and anti-rabbit antibodies (Bio-Rad Laboratories, Hercules, CA, USA) diluted 1:1,000 to 1:2,000 in 1% fat-free milk in TBS-T at room temperature for 1 hours. Detection was achieved using Supersignal West Pico Chemiluminescent Substrate (ThermoFisher Scientific Inc., Rockford, IL, USA). Quantitation of Western blots by densitometry was performed with Genetool version 4.03.05 (SynGene, Cambridge, England).
Statistical analysis
Data are expressed as mean±standard error of the mean (SEM). Differences between groups were evaluated using SigmaPlot version 13.0 (Systat Software Inc., San Jose, CA, USA). The independent t test and one-way analysis of variance were used to analyze the quantitative variables between groups. A P<0.05 was deemed to indicate significance.
RESULTS
Gemigliptin reduced DPP-IV in ECs
In HUVECs, DPP-IV protein levels were increased in a dose-dependent manner following IL-1β treatment (Supplemental Fig. S1A, B). HUVECs were stimulated with 10 ng/mL of IL-1β for various times (0, 5, 15, 30, 60 minutes, or 24 hours), and DPP-IV protein levels were significantly increased after 15 minutes of treatment (Supplemental Fig. S1C, D). IL-1β-induced GLP-IR protein levels were decreased in a dose-dependent manner, and then decreased at 24 hours after IL-1β treatment (Supplemental Fig. S1A–D). In contrast, DPP-IV protein levels were increased in a dose-dependent manner following gemigliptin treatment (Supplemental Fig. S1E, F). When HUVECs were stimulated with gemigliptin, DPP-IV protein levels decreased in a time-dependent manner (Supplemental Fig. S1G, H). In addition, gemigliptin-induced GLP-1R protein levels were increased in a dose- and time-dependent manner (Supplemental Fig. S1E–H). Gemigliptin treatment significantly reduced IL-1β-induced DPP-IV protein expression in ECs (Supplemental Fig. S2). Furthermore, GLP-IR protein levels were increased following treatment with 20 μM gemigliptin for 24 hours (Supplemental Fig. S2).
Gemigliptin inhibits IL-1β-induced EndMT and conserves the endothelial phenotype and migration
We assessed the effects of gemigliptin on IL-1β-mediated EndMT in cultured ECs. Exposure of HUVECs to IL-1β for 1 day triggered an obvious alteration in cellular morphology, from a cobblestone-like shape to a more spindled, fibroblast-like shape (Supplemental Fig. S3A). In contrast, gemigliptin treatment significantly suppressed this transition by inverting the endothelial phenotype (Supplemental Fig. S3A). To examine the effects of gemigliptin on IL-1β-mediated EndMT, we assessed the presence of fibroblast cell marker α-SMA and EC marker vWF by immunofluorescence (Supplemental Fig. S3B). Representative immunostaining revealed that IL-1β-mediated EndMT was associated with reduced vWF levels (Supplemental Fig. S3C) and increased α-SMA levels (Supplemental Fig. S3C). However, upon gemigliptin treatment, most cells were positive for vWF and negative for α-SMA (Supplemental Fig. S3B).
Furthermore, we examined the effects of IL-1β and gemigliptin on chemotactic motility using a transwell chamber. IL-1β treatment resulted in a significantly reduced number of migratory cells in HUVECs (Supplementary Fig. 3D, E). Consistent with the results of the Transwell assays, the migration rates were significantly higher after gemigliptin treatment (Supplemental Fig. S3D, E). We examined the effect of gemigliptin on wound healing using a HUVEC monolayer. Wound healing cell invasion assays revealed that IL-1β also inhibited the migration of fibroblast-like EndMT cells, while gemigliptin induced the invasion of these cells (Supplemental Fig. S3F). Thus, gemigliptin stimulated migration of HUVECs.
Gemigliptin inhibits IL-1β induction of SMC markers and MSC markers, but reverses EC marker expression
These observations were further confirmed by Western blotting. In the presence of IL-1β, eNOS, VE-cadherin, and vWF protein levels were reduced, and α-SMA and SM22 protein levels, SMC markers, were increased (Fig. 1). The mesenchymal markers FSP-1, FN, and Col I were increased after IL-1β treatment (Fig. 1). However, gemigliptin treatment reversed these IL-1β-mediated effects. Quantitative analysis of the Western blots verified our results. These findings demonstrate that gemigliptin suppresses EndMT induced by IL-1β.
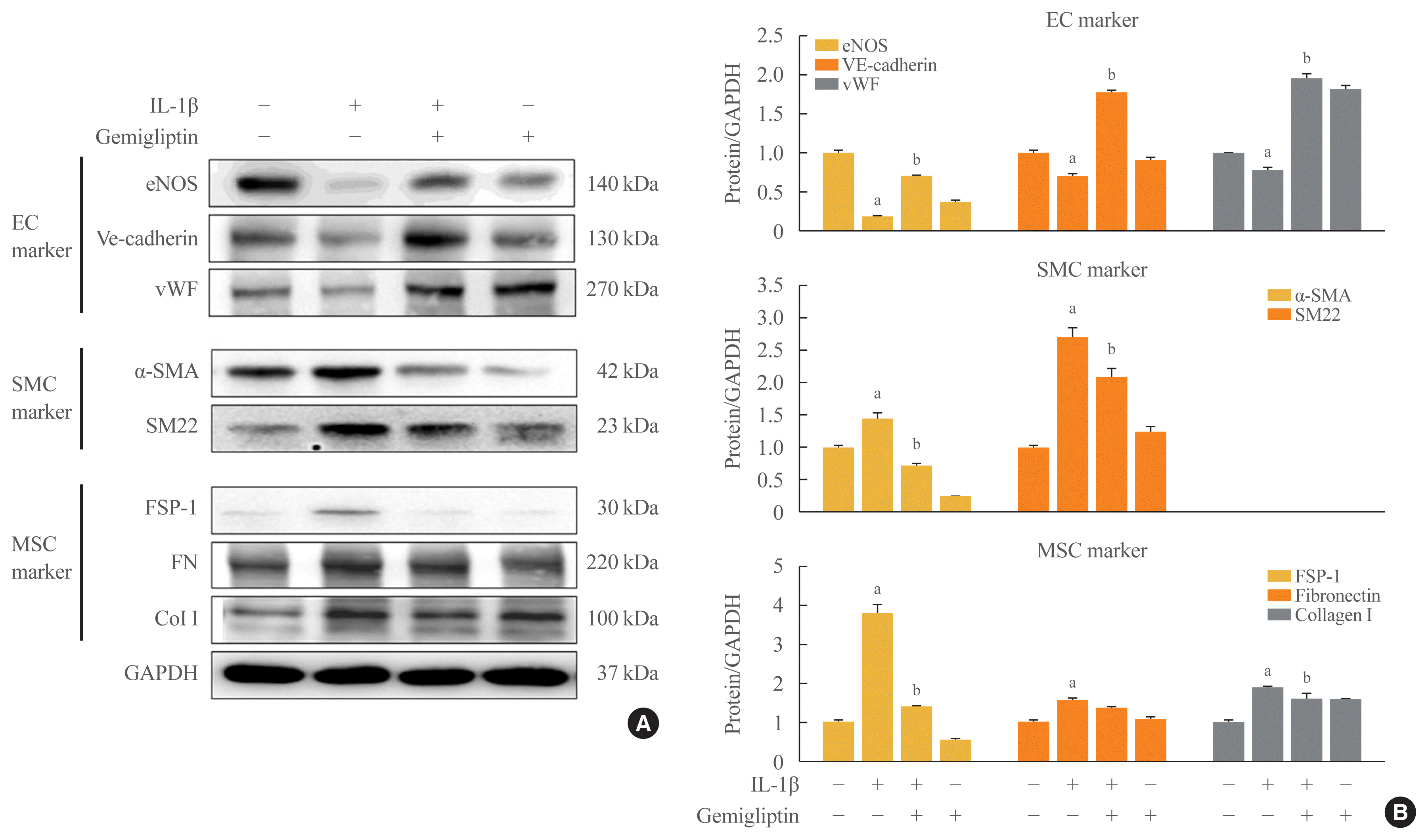
Fig. 1
Gemigliptin significantly but not completely suppressed the interleukin-1β (IL-1β)-induced smooth muscle cell (SMC) markers and mesenchymal stem cell (MSC) markers, reversed endothelial cell (EC) markers. Human umbilical vein endothelial cells (HUVECs) were treated IL-1β (10 ng/mL) in the presence of absence of 20 μM gemigliptin for 24 hours. (A) Representative Western blot image comparing changes in the expression of the endothelial cell marker endothelial nitric oxide synthase (eNOS), vascular endothelial cadherin (VE-cadherin), von Willebrand factor (vWF), the smooth muscle marker α-smooth muscle actin (α-SMA), transgelin (SM22), and mesenchymal cell marker fibroblast-specific protein 1 (FSP-1), fibronectin (FN), and collagen type I (Col I). (B) Quantification of protein expression by densitometry analysis of Western blots. Results were normalized with glyceraldehyde 3-phosphate dehydrogenase (GAPDH; loading control) and expressed as fold-change relative to control without IL-1β. Values are mean±standard error of the mean (n=3). aP<0.05, control vs. IL-1β; bP<0.05, IL-1β vs. IL-1β+gemigliptin.
![]()
Gemigliptin inhibits IL-1β induction of the BMP signaling pathway
To investigate the mechanism by which IL-1β is associated with BMP activity, key changes in BMP signaling were detected by Western blotting. We investigated BMP signaling during IL-1β-mediated EndMT. After IL-1β treatment of HUVECs for 24 hours, BMP2 and BMP7 were significantly increased, whereas BMP4 was suppressed (Fig. 2A, B). Because the biological functions of BMP2 and 7 are mediated through BMP/activin signaling via the six BMP- and activin-specific receptors, Act-RIA, Act-RIIA and RIIB, BMP-RIA and RIB, and BMP-RII, we analyzed the expression of these receptors after 24-hour IL-1β treatment in HUVECs. The expression of Act-RIA and BMP-RIB, including BMP receptor type I, significantly increased, and Act-RIIA, Act-RIIB, and BMP-RII, including BMP receptor type II, were activated by IL-1β (Fig. 2A, B). Gemigliptin significantly inhibited the effects of IL-1β on Act-RIA and BMP-RIA protein levels and Act-RIIB and BMP-RII protein expression (Fig. 2A, B). Furthermore to investigate the mechanism by which IL-1β is associated with TGF-β1, key marker in EndMT, we were detected by Western blotting. After IL-1β treatment of HUVECs for 24 hours, TGF-β1 and TGFβ-RI protein levels were increased, whereas TGF β-RII protein levels were decreased (Fig. 2C, D). Gemigliptin significantly inhibited the effects of IL-1β on TGFβ-RII protein levels. Therefore, gemigliptin blocks the IL-1β-induced BMP signaling pathway.
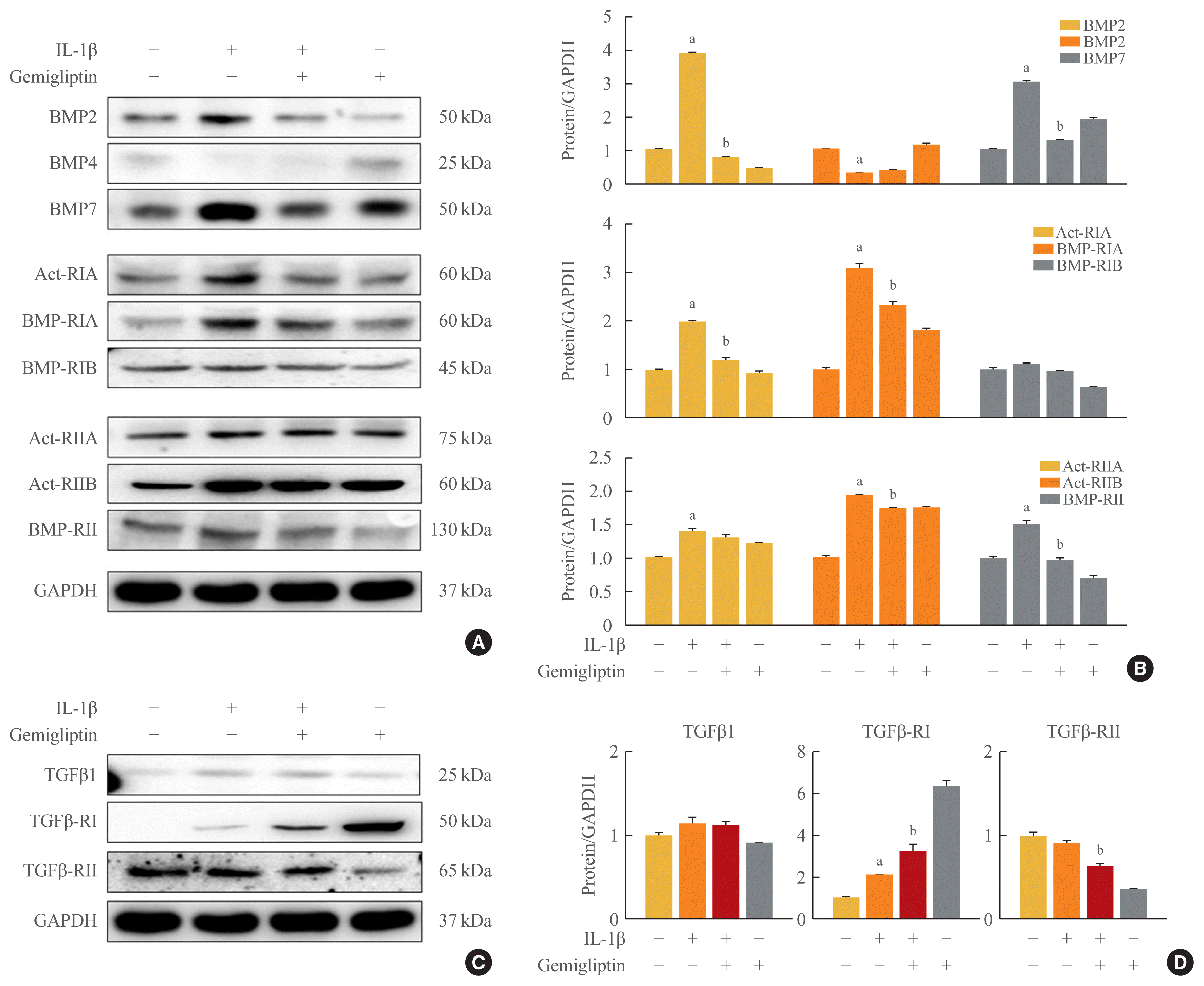
Fig. 2
Gemigliptin inhibits interleukin-1β (IL-1β)-induced the bone morphogenetic protein (BMP) signaling pathway. Human umbilical vein endothelial cells (HUVECs) were treated IL-1β (10 ng/mL) in the presence of absence of 20 μM gemigliptin for 24 hours. (A) Representative Western blot image comparing changes in the expression of the BMP2, BMP4, BMP7 and activin receptor type IA (Act-RIA), activin receptor type IIA (Act-RIIA), and activin receptor type IIB (Act-RIIB), and BMP receptor type IA (BMP-RIA) and IB (BMP-RIB), and type II (BMP-RII). (B, D) Quantification of protein expression by densitometry analysis of Western blots. (C) Representative Western blot image comparing changes in the expression of the transforming growth factor β1 (TGF-β1) and TGF-β receptor type I (TGF-β-RI), and type II (TGF-β-RII). Results were normalized with glyceraldehyde 3-phosphate dehydrogenase (GAPDH; loading control) and expressed as fold-change relative to control without IL-1β. Values are mean±standard error of the mean (n=3). aP<0.05, Control vs. IL-1β; bP<0.05, IL-1β vs. IL-1β+gemigliptin.
![]()
Gemigliptin inhibits IL-1β-induced Smad-dependent pathway
The binding of BMP to BMP receptors induces heterodimeric complexes and subsequently activates Smad or mitogen-activated protein kinases (MAPKs) by phosphorylation [14]. We found that the Smad1/5/9, Smad2, and Smad3 signaling pathways were activated by IL-1β. IL-1β significantly increased the phosphorylation of Smad1/5/9, Smad2, Smad3, and endogenous Smad2 and Smad3 (Fig. 3). Gemigliptin-treated cells showed a significant reduction in the phosphorylation of Smad1/5/9, Smad2, and Smad3 induced by IL-1β. Densitometric analyses of band intensities showed that gemigliptin partially increased endogenous Smad1/5/9 in contrast to Smad2 and 3 (Fig. 3). These results indicate that gemigliptin blocks IL-1β-induced EndMT in part by inhibiting the BMP/Smad signaling pathway.
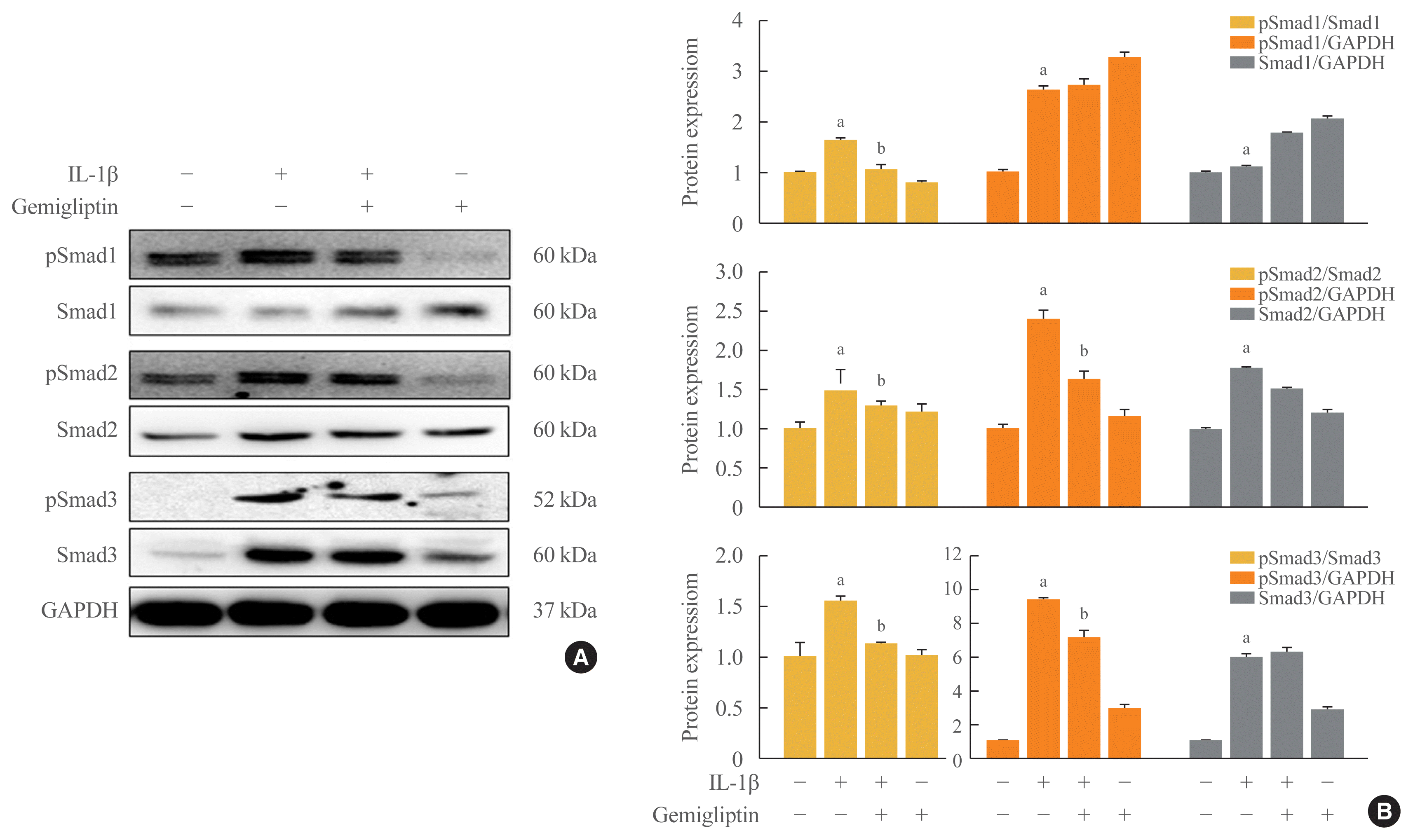
Fig. 3
Gemigliptin inhibits interleukin-1β (IL-1β)-induced the Smad-dependent pathway. Human umbilical vein endothelial cells (HUVECs) were treated IL-1β (10 ng/mL) in the presence of absence of 20 μM gemigliptin for 24 hours. (A) Representative Western blot image comparing changes in the expression of the Smad1, Smad2, Smad3 and the phosphorylation of Smad1 (pSmad1), Smad2, Smad3. (B) Quantification of protein expression by densitometry analysis of Western blots. Results were expressed as fold-change relative to control without IL-1β. Values are mean±standard error of the mean (n=3). GAPDH, glyceraldehyde 3-phosphate dehydrogenase. aP<0.05, Control vs. IL-1β; bP<0.05, IL-1β vs. IL-1β+gemigliptin.
![]()
Gemigliptin inhibits IL-1β-induced non-Smad BMP pathway
Because inflammatory cytokines are primarily associated with the activation of MAPK signaling, we tested whether gemigliptin regulates IL-1β-induced EndMT through this pathway. We treated HUVECs with IL-1β or gemigliptin for 0, 15, 30, 60 minutes, and 24 hours. We observed that the addition of IL-1β to the culture medium induced a rapid and strong activation of MAPK signaling, including Erk, p38, and JNK in HUVECs (Supplemental Fig. S4). After the addition of gemigliptin, only Erk was phosphorylated (Supplemental Fig. S4). The Erk, p38, and JNK phosphorylation via IL-1β-induced EndMT was strongly inhibited in the cells treated with gemigliptin (Fig. 4). Therefore, our results indicated that IL-1β upregulated MAPK activity, but the effect was prevented by gemigliptin.
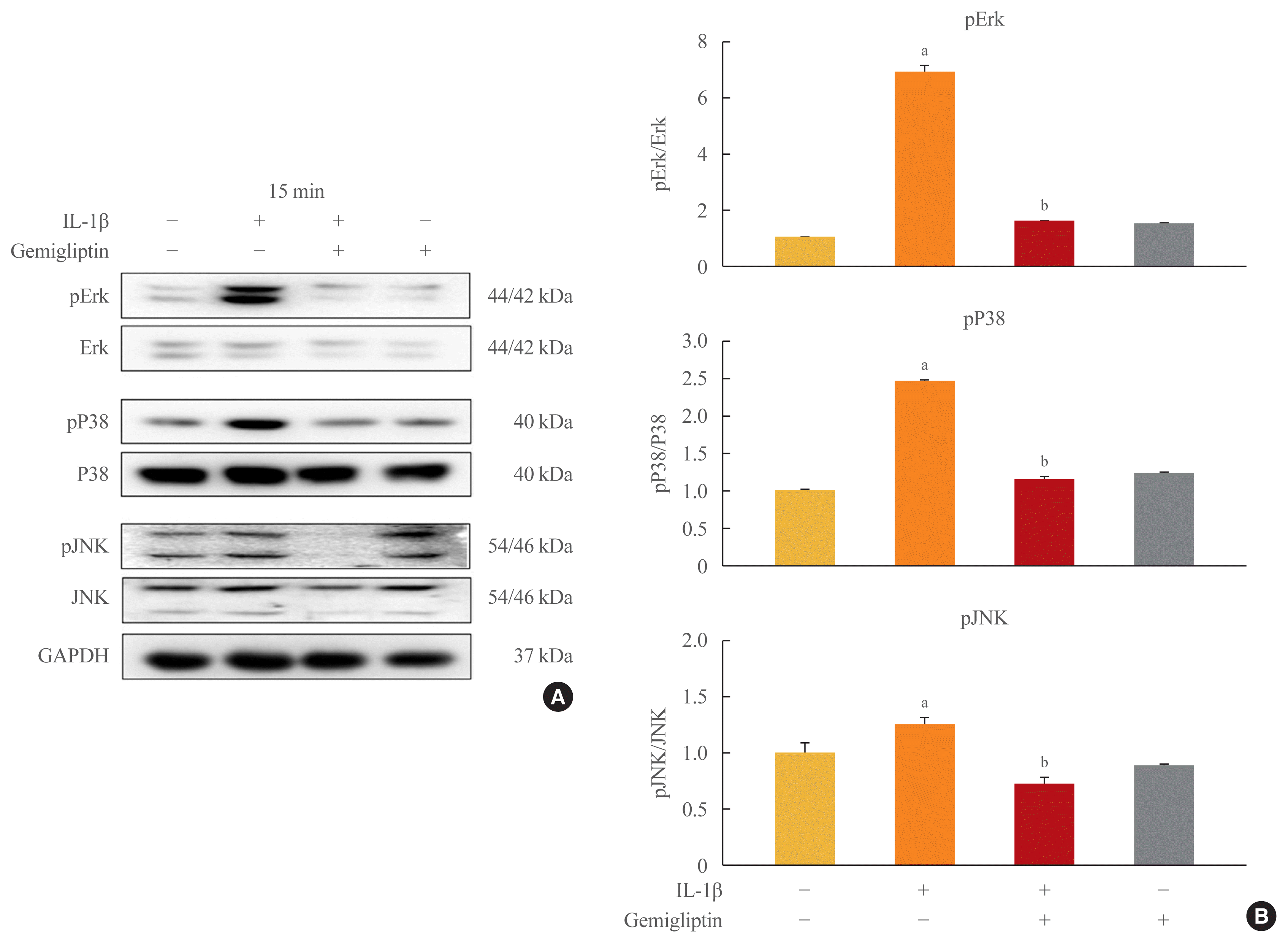
Fig. 4
Gemigliptin inhibits interleukin-1β (IL-1β)-induced the non-Smad bone morphogenetic protein pathway. Human umbilical vein endothelial cells were treated IL-1β (10 ng/mL) in the presence of absence of 20 μM gemigliptin for 15 minutes. (A) Representative Western blot image comparing changes in the expression of the extracellular regulated protein kinase (Erk), P38, JNK and the phosphorylation of Erk (pErk), P38 (pP38), JNK (pJNK) after exposure to treated IL-1β (10 ng/mL) in the presence of absence of 20 μM gemigliptin for 15 minutes. (B) Quantification of protein expression by densitometry analysis of Western blots. Values are mean±standard error of the mean (n=3). GAPDH, glyceraldehyde 3-phosphate dehydrogenase. aP<0.05, Control vs. IL-1β; bP<0.05, IL-1β vs. IL-1β+gemigliptin.
![]()
Gemigliptin inhibits IL-1β-induced osteogenic-specific transcription factors
Because of the positive role of BMP/Smad signaling in regulating Runx2 expression, we examined the effects of IL-1β-activated BMP2 on osteogenic-specific transcription factors Runx2, osterix, and hepcidin in HUVECs. The osterix, Runx2, and hepcidin transcriptional activities tended to increase when HUVECs were treated with 10 ng/mL IL-1β, even though in the presence of gemigliptin their significantly suppressed (Fig. 5).
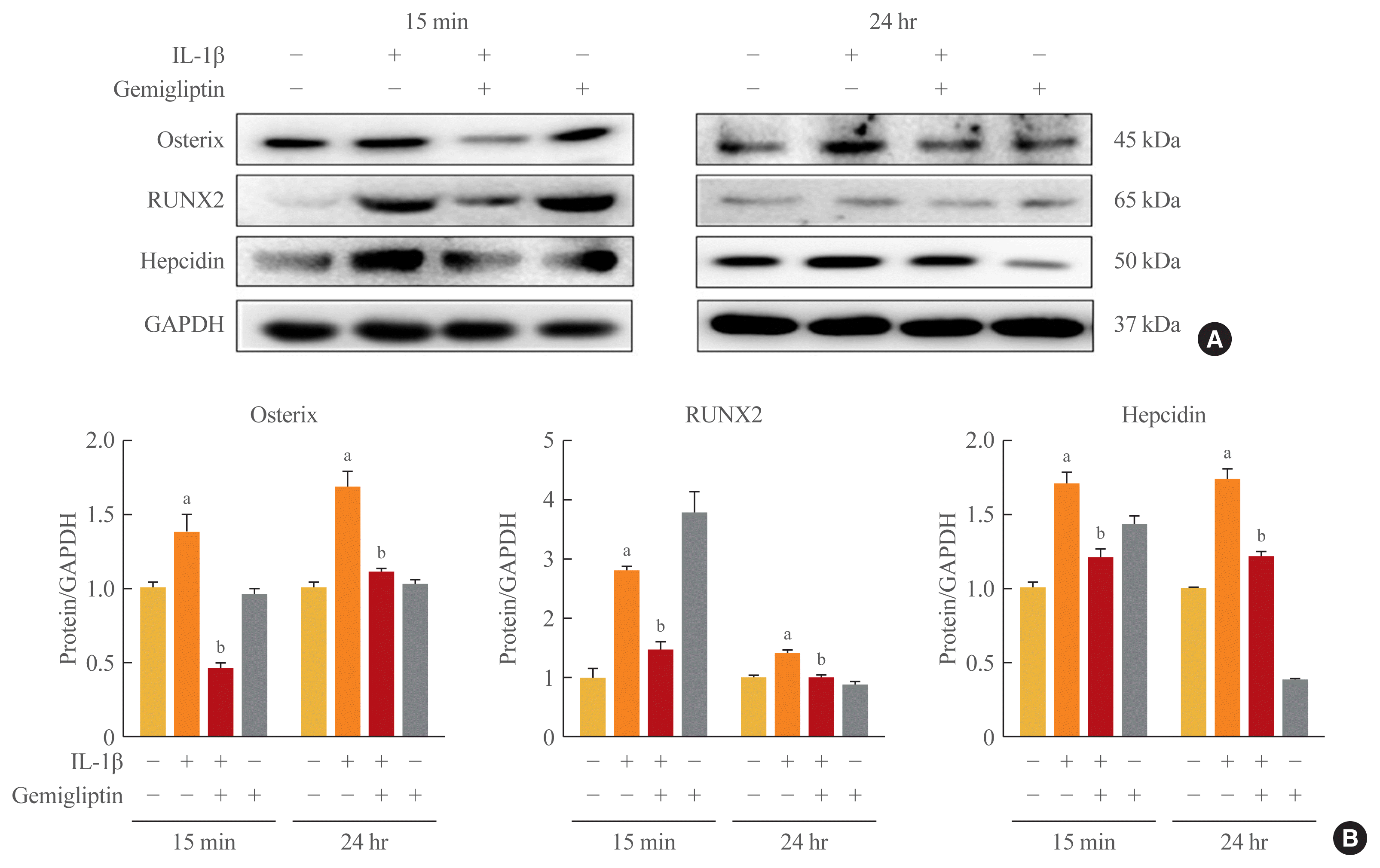
Fig. 5
Gemigliptin inhibits interleukin-1β (IL-1β)-induced osteogenic-specific transcription factor. Human umbilical vein endothelial cells were treated IL-1β (10 ng/mL) in the presence of absence of 20 μM gemigliptin for 15 minutes or 24 hours. (A) Representative Western blot image comparing changes in the expression of the osterix, runt-related transcription factor 2 (RUNX2), and hepcidin. (B) Quantification of protein expression by densitometry analysis of Western blots. Results were normalized with glyceraldehyde 3-phosphate dehydrogenase (GAPDH; loading control) and expressed as fold-change relative to control without IL-1β. Values are mean±standard error of the mean (n=3). aP<0.05, Control vs. IL-1β; bP<0.05, IL-1β vs. IL-1β+gemigliptin.
![]()
DISCUSSION
The EndMT is associated with a number of diabetic complications and has been shown to contribute to diabetic cardiomyopathy, diabetic nephropathy, diabetes-associated kidney fibrosis, and diabetic retinopathy through various signaling pathways [4,15]. To the best of our knowledge, this study is the first to demonstrate that IL-1β induces the EndMT in HUVECs, and that this pathological process may be inhibited by gemigliptin, a new DPP-IV inhibitor.
In many studies, cardiovascular diseases such as hypertension, atherosclerosis, and heart failure have been associated with abnormal vascular remodeling following vascular damage to fundamental interactions, ECs, and VSMCs [1,2]. Gemigliptin (brand name: Gemiglo), developed by LG Chem., is a potent, selective, competitive, and long-acting DPP-IV inhibitor. Choi et al. [12] reported that gemigliptin directly attenuates the abnormal proliferation and migration of primary VSMCs, and endogenous DPP-IV in VSMCs contributed to the effects of gemigliptin. Gemigliptin induced HUVEC proliferation in a dose-dependent manner (data not shown), and linagliptin in particular prevented aortic and endothelial stiffness regardless of its glycemic-lowering effects [16]. Fig. 1 shows that endogenous DPP-IV in HUVECs is suppressed by gemigliptin, but GLP-1R is not. Because GLP-1R activates the inflammatory response and enhances angiogenesis during the early proliferative phase of wound healing [17], increased GLP-1R expression was an initial cellular reaction after IL-1β or gemigliptin treatment.
The EndMT is the process by which ECs progressively lose endothelial features and gain mesenchymal fibroblast-like characteristics, transitioning from a differentiated to an undifferentiated state [1,2]. Phenotypic changes observed in the EndMT are also accompanied by cytoskeletal restructuring, which alters EC polarity, giving rise to a morphology commonly described as spindle-shaped and fibroblast-like [1,2]. Multiple mesenchymal markers are used to characterize the EndMT, including the myogenic proteins, α-SMA, SM22, and calpoin, and non-myogenic fibroblast markers such as vimentin, FSP-1 and several collagens [3,4]. During kidney fibrosis, the endothelial origin of up to 50% of all fibroblasts is observed by detecting the co-expression of the endothelial marker, CD31, and the fibrotic marker, FSP-1 [4]. Co-expression of smooth muscle markers in ECs has been described previously, and freshly purified adult bovine arterial ECs have shown spontaneous and progressive increases in the expression of smooth muscle markers over time in culture [18]. The α-SMA and SM22 are considered early markers of SMCs, and SM-myosin heavy chain is a late marker of SMCs [19]. Our results showed that the FN and Col I proteins were expressed in control cells in the absence of IL-1β treatment (Fig. 1). Proteins that contribute to the ECM, including FN, Col I, Col III, and vimentin, increase during cell proliferation and aging [19]. It is possible that our findings indicated an intermediate phenotype of the EndMT, which has a partial phenotypic transition, because loss of the endothelial phenotype during transition has been proposed to occur after the completion of EndMT [18].
Maleszewska et al. [6] demonstrated that IL-1β acts synergistically in the presence of TGF-β2 to induce the EndMT in HUVECs, and TGF-β2 is also progressively upregulated during the EndMT. IL-1β is a more potent inducer of the EndMT than TGF-β and TNF-α [3]. Most BMPs promote the EndMT; recombinant BMP4 treatment induced the EndMT in HUVECs and human cutaneous microvascular endothelial cells [20], and BMP7 treatment appears to be a negative regulator of the EndMT [10]. Cohen [21] suggested that IL-1β does not activate the BMP signaling pathway directly, whereas Shanmugam et al. [22] reported a novel connection between IL-1β and the activation of BMP signaling in human hepatocytes. IL-1β increased BMP-2 mRNA levels 6- and 3-fold in islets of Langerhans isolated from neonatal rats and humans, respectively [23], whereas BMP4 expression was reduced. BMP4 is expressed at about 1/100 the level of BMP2, and unlike BMP2, it is not upregulated by IL-1β in human hepatocytes. Therefore, IL-1β activated BMP2 expression, and BMP4 was not present in HUVECs (Fig. 2).
Cho et al. [2] suggested that the inflammation-induced EndMT was largely controlled by the TGFβ signaling and non-TGFβ signaling pathways. Despite TGFβ being the most well-known EndMT inducer [2,3], activation of the TGFβ signaling pathway by IL-1β is not significant compared with that of the BMP signaling pathway (Fig. 2). Many studies have shown that each EC subtype responds differently to different inflammatory stimuli in vitro [2,8]. Endothelial heterogeneity was studied in relation to the molecular mechanisms and functions associated with EndMT processes; especially, IL-1β-stimulated organ-specific endothelial heterogeneity has been reported [8]. Further studies are needed to completely elucidate the mechanisms associated with TGF-β expression via IL-1β-induced EndMT.
The BMP2 signaling cascade is initiated by activation of BMP-RIA (ALK3), BMP-RIB (ALK6), and BMP-RII [20]. Our results indicated that the BMP type I receptor (ALK3) and BMP type II receptor (BMPR-II, Act-RIIB) activated the IL-1β-induced EndMT, and BMP receptor suppression led to decreased Smad1 phosphorylation by gemigliptin treatment. BMP2 signaling was shown to inhibit IL-1 receptor signaling through R-Smads (i.e., Smad1/5/8 and Smad2/3). Because BMP2 and BMP4 share downstream signaling pathways, suppression of Smad1/5/8 and Smad2 by gemigliptin may result in BMP2 inhibition of proinflammatory cytokines and anti-inflammatory molecules. ALK2 suppression by gemigliptin can decrease Smad2/3 signaling. IL-1β has been identified as a TGF-β/Smad target gene [24]. As shown in Fig. 4, the MAPK signaling pathway was increased after 24 hours; this was thought to be due to the continuation of EC growth induced by gemigliptin, because the EC marker, vWF, was remarkably increased by treatment with gemigliptin only (Fig. 1). In our experiments, the MAPK pathway showed very rapid signaling, whereas SMAD signaling had a very long duration. Ultimately, these data indicate that gemigliptin inhibits BMP2 signaling via Smads in the IL-1β-induced EndMT.
The downregulation of endothelial markers and upregulation of fibrotic proteins is tightly controlled at the genetic level by several transcription factors. Inflammatory cytokines are primarily associated with the activation of MAPK signaling, and Runx2 is activated via several of the MAPK pathways [25]. BMP2 activates the p38, Erk1/2, and JNK1/2 signaling pathways to promote the expression and activation of Runx2. BMP2 modulates osteoblastic differentiation through the canonical BMP/Smad pathway and non-canonical BMP pathway [26]. IL-1β can activate the BMP signaling pathway by increasing the expression of BMP2, and is correlated with IL-1β-induced upregulated hepcidin [22]. We did not demonstrate that Forkhead Box M1 (Foxm1) transcription factor (FOXM1) activation of activating transcription factor 3 (ATF-3) is a driver of the TGF-β-induced EndMT in ECs. Furthermore, Foxm1 bound to and increased the promoter activity of the Snail gene, which encodes a critical transcriptional regulator of the EndMT [27]. ATF-3 responds to TGF-β and controls the expression of the primary epithelial-to-mesenchymal transition markers, Snail, Slug, and Twist [28]. These three transcription factors are thought to be associated with TGF-β-induced EndMT. The activation of BMP2 via IL-1β-induced EndMT promotes the expression and activation of Runx2, Osterix, and hepcidin, and gemigliptin inhibits the activation of MAPKs via IL-1β.
DPP-IV inhibitor has unique drug-specific effects and has been observed to have various effects on endothelial biology [29]. Our results showed that vWF and BMP7 were significantly increased by gemigliptin alone. BMP7 plays an opposing role to TGF-β1 in terms of cell proliferation. BMP7 treatment also exerted an anti-inflammatory effect [30–32]. The DPP-IV inhibitor has an anti-apoptotic effect and potential anti-atherosclerotic properties [33]. It is possible that potentiation of BMP7 by DPP-IV inhibitors may enhance the inflammatory and proliferative responses to IL-1β, which may in turn aggravate the course of endothelial transition. DPP-IV is essential for TGF-β-induced receptor hetero-dimerization and subsequent intracellular signal transduction. Protein-protein interactions of TGFβ receptor I and II are critical for TGF-β signal transduction [29]. Nevertheless, gemigliptin increased TGFβ-RI expression without activation of TGF-β-RII (Fig. 2C, D). Gemigliptin did not increase TGF-β1 (Fig. 2C, D), and it is thought to be a reaction of TGFβ-RI by gemigliptin.
In conclusion, gemigliptin inhibited the IL-1β-induced EndMT differentiation of HUVECs via BMP2/Smad and MAPK/p38 pathways. Also osteoblastic markers were observed to reduce the IL-1β-induced EndMT differentiation of HUVECs.
 XML Download
XML Download