

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Acromegaly is a chronic endocrine disease characterized by growth hormone (GH) elevation, >95% of the time due to hypersecretion by somatotroph cells of a benign pituitary neoplasm, or a somatotropinoma [1]. Prolonged excess GH causes a plethora of symptoms, stimulating the liver to secrete more insulin-like growth factor 1 (IGF-1) and generating musculoskeletal changes, like acral enlargement, increased skin thickness, soft tissue hypertrophy, and facial bony deformities may become apparent in such patients [2,3]. At diagnosis most patients present with varying systemic comorbidities: diabetes mellitus, hypertension, cardiomyopathy, valve disorders, colon polyps, goitre, carpal tunnel syndrome, sleep apnea, and sexual disturbances [1]. Local tumor mass effects may lead to visual impairment, hypopituitarism, and headache [1]. Cardiovascular, metabolic and oncologic diseases act as principal factors for the estimated 1.5 to 2.5 standardized mortality ratio in patients with acromegaly [3–6].
Albeit a rare disease, population-based studies have been able to determine the prevalence, ranging from 28 to 134 cases per million (cpm) and the incidence, ranging from 1.9 to 11 cpm per year [7–19]. Median age at diagnosis is within the fifth decade of life, but owing to the small population sizes it is hard to infer differences in gender [20,21]. Majority of the epidemiological data has come from Europe [7,9–12,14,16–18], but some representative countries, like South Korea in Asia have also made such contributions via multi-center [13] and claims database studies [15]. Incidence of acromegaly has been on the rise, potentially due to improved diagnostic tools, disease perception among physicians and the public, and a real increase in prevalence [21]. Despite these developments, prognosis is harmed with a median diagnostic delay of 4.5 to 5 years and associated comorbidities [21].
Late diagnosis and management are a hurdle that necessitates the initiation of precision therapy, or customizing treatment to specific patients, in everyday practice. Surgery, acting as first line, followed by medical therapy (somatostatin analogues [SSAs], GH receptor antagonists, and dopamine agonists [DAs]), radiotherapy, radiosurgery (gamma knife surgery), and different combinations of each have been shown to result in a decrease in morbidity and mortality to general population [1,22]. However, cases of resistance to surgical or medical treatment require an integration of clinical and molecular predictors to aid in selecting the best tailored treatment. Simultaneous consideration of all predictors, such as the patient’s gender, age, pre-operative hormonal levels, tumor size, somatostatin and dopamine receptor sensitivity and expression, and signaling pathway disruption, among others plays a role in improving remission rates [23–38].
This review compares current data from epidemiological studies to judge the reality of precision therapy with a special focus on its relevance to the South Korean population.
Go to : 
EPIDEMIOLOGY IN KOREA AND THE WORLD
To this day, epidemiological data of acromegaly is highly variable, as shown by estimations of 20 studies in Table 1. Prevalence of acromegaly ranges from 18 to 133 cpm, whereas the incidence ranges from 1.9 to 11 cpm/year [7–19,39–45].
Table 1
| Study | Country | Median age at diagnosis, yr | Males: Females | Population covered | Cases | Years of study | Duration, yr | Macroadenomas, % | Annual incidence rate, cpm/yr | Prevalence rate, cases/million | Type of study |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Asia | |||||||||||
| Kwon et al. [13], Park et al. [15] | Korea | 44.1 | 627:723 | 48,000,000 | 718/1,350 | 2003–2007/2010–2013 | 4/study | 83 | 3.6 & 3.9 | 28 | Referral/claims database |
| Khamseh et al. [39] | Iran | 44 (mean) | 47:38 | NA | 85 | 2014–2016 | 3 | 73 | NA | NA | National registry |
| Tseng et al. [40] | Taiwan | 41.8 (mean) | 117:155 | NA | 272 | 2013–2015 | 3 | 70 | NA | NA | National registry |
|
|
|||||||||||
| North America | |||||||||||
| Burton et al. [8] | United States | 41 (mean) | Equal | 50,000,000 | 2,241 | 2008–2012 | 5 | NA | 11 | 78 | Claims database |
| Portocarrero-Ortiz et al. [19] | Mexico | 41 (mean) | 1,007:1,050 | 119,000,000 | 2,057 | 2009–2012 | 4 | 74 | NA | 18 | National registry |
| Vallette et al. [45] | Canada | 45 | 329:320 | NA | 649 | 1980–2010 | 31 | 79 | NA | NA | Referral centers |
|
|
|||||||||||
| Nordic Europe | |||||||||||
| Tjornstrand et al. [18] | Sweden | 51.7 | 27:26 | 1,600,000 | 53 | 2001–2011 | 11 | 65 | 3.5 | NA | National registry |
| Raappana et al. [16] | Finland | 40.5 | 41:38 | 722,000–733,000 | 40 | 1992–2007 | 16 | 78 | 3.4 | NA | Referral centers |
| Dal et al. [10] | Denmark | 48.7 | 214:191 | 7,200,000 | 405 | 1991–2010 | 20 | 69 | 3.8 | 85 | National registry |
| Hoskuldsdottir et al. [12] | Iceland | 45 (mean) | NA | 316,075 | 52 | 1955–2013 | 59 | 78 | 8 | 133 | Referral centers |
|
|
|||||||||||
| Non-Nordic Europe | |||||||||||
| Caputo et al. [9] | Italy | 50.9 | 146:223 | 4,400,000 | 369 | 2012–2016 | 5 | NA | 5.3 | 83 | Claims database |
| Mestron et al. [14] | Spain | 45 (mean) | 478:741 | Spain population 2001 | 1,219 | 1997–2004 | 8 | 69 | 2.1 | 34 | National registry |
| Gruppetta et al. [11] | Malta | 44 | 22:30 | 417,608 | 52 | 2000–2011 | 12 | 73 | 3.1 | 125 | Referral center |
| Bex et al. [7] | Belgium/Luxembourg | 42–46 | 213:205 | 10,850,000 | 418 | 2000–2004 | 5 | 79 | 1.9 | 59 | Referral centers |
| Reincke et al. [17] | Germany | 44 (mean) | 709:833 | 80,000,000 | 1,543 | 2003–2005 | 3 | 81 | NA | 30 | National registry |
| Fernandez et al. [42] | UK | 47 | 4:3 | 81,149 | 7 | 2006 | NA | 86 | NA | 86 | Multi-center, cross-sectional |
| Maione et al. [43] | France | 46 | 460:539 | NA | 999 | 1977–2012 | 35 | 67 | NA | NA | National registry |
| Daly et al. [41] | Belgium | 47 | 6:3 | 71,962 | 9 | 2005 | NA | 89 | NA | 125 | Multi-center, cross-sectional |
| Petrossians et al. [44] | EU Multicenter (10 countries) | 45 | 1,444:1,729 | NA | 3,173 | 2012–2015 | 4 | 72 | NA | NA | Referral centers |
![]()
Highest prevalence of 133 cpm was reported in Iceland, while 125 cpm was reported in Malta and Belgium both [11,12,41]. The sample size in the three studies and the catchment population were lower compared to other studies, like in two North American studies by Burton et al. [8] and Portocarrero-Ortiz et al. [19], which presented over 2,000 cases each and yet, published lower prevalence rates. Moreover, the cases in Iceland and Malta were compiled over 59 and 12 years periods, respectively, accumulating cases and leading to higher perceived prevalence to previously reported studies of shorter length [11,12]. Interestingly, despite having one of the largest sample sizes, the Mexican national registry reports the lowest prevalence of 18 cpm, even lower than previously reported data by Kwon et al. [13] (28 cpm), Reincke et al. [17] (30 cpm), and Mestron et al. [14] (34 cpm) [19]. Colao et al. [1] argued that the increasing prevalence in newer studies could be due to improvements in the diagnostic approach, or to the availability of larger databases. The former can be explained by higher rates of economic growth, improvement and access to healthcare [15]. The latter may be inaccurate, as the largest databases present in this review have lower prevalence rates than in Iceland or Malta and, yet have discrepancy amongst each other, potentially due to difference in data collection methods [8,19]. Most studies are 3 to 5 years in length and if prolonged, could showcase a higher prevalence rate. Actual prevalence of this disorder may be higher in the rest of the studies, owing to increased patient survival and developments in screening, surgical practice, post-operational follow-up, and medical treatment [1].
Furthermore, the highest incidence rate of 11 and 8 cpm/year were reported by Burton et al. [8] and Hoskuldsdottir et al. [12], respectively, while the majority of other studies present an incidence rate ranging from 3 to 4 cpm/year [10,11,13,15,16,18]. Improved awareness among internists and general practitioners, rise of social media and the introduction of a universal diagnostic method possibly lead to the increase in incidence rates in recent studies [1,46]. The lowest incidence was reported by Bex et al. [7] (1.9 cpm/year) and Mestron et al. [14] (2.1 cpm/year). Innovative methods have been recently published by Burton et al. [8] and Park et al. [15] about using health insurance data and national claims database for the estimation of incidence rates. Two 4-year studies, by Kwon et al. [13] (2003 to 2007) and Park et al. [15] (2010 to 2013) presented comparable incidence rates of 3.6 and 3.9, respectively, which solidified the incidence rate of acromegaly in South Korea. As far as this review is concerned, this nation is the only one that has implemented varying data collection methods (referral centers and claims database) in checking the accuracy of its epidemiological data for this disease.
Median age at diagnosis in South Korea is 44.1, following the 40.5 to 51.7 age range in other studies to date (Table 1) [7–21,39–46]. It is difficult to establish if there is a difference in prevalence of either gender, as there are 10 studies showing a higher prevalence in women, including Korea [9,11,13–15,17, 19,40,43,44], and eight, showcasing the opposite [7,10,16,18, 39,41,42,45]. Generally, this could indicate an equal occurrence in either gender (Table 1) [7–21,39–46]. One of the major contributors to high morbidity in acromegaly is the delay in diagnosis, which according to recent studies is approximately 5 years [12,14,21]. Clinical characteristics and overall comorbidity incidence have been similar among Korean and Western studies [1,2,15], but the former showed that females were at higher risk of malignancy [2,15]. This also necessitates more widespread research on geographical differences in acromegaly-associated comorbidities.
Seeing as how there is insufficient epidemiological data in non-European countries, there is a need for international studies, making a modern comparison of geographical variation, ethnicity, sex, healthcare system type, and access to healthcare. Establishment of Liege Acromegaly Survey Database across 14 centers in Europe has been exemplary of international cooperation in epidemiological research, presenting the largest sample size to date [44]. Before other continents follow Europe’s footsteps, more population-based studies or databases need to be set up within nations. North American [8,19,45] or Nordic European [10,12,16,18] countries could be next to follow and pool resources, despite each country implementing varying data collection methods. In regards to Asia, Korea has completed two different types of population-based studies, which have yielded similar incidence results. Data about newly established national pituitary tumor registries in Iran and Taiwan have been published [39,40]. Their future use, if updated with prevalence and incidence data, and the appearance of new registries in more countries will be a great step for the realization of any ethnic and geographical comparisons.
Go to :
CURRENT TREATMENT APPROACHES
Historically, surgical excision of pituitary adenomas in a transsphenoid surgery (TSS) has been the first-line treatment, being the only modality able to reduce morbidity and mortality and cause immediate hormonal normalization [1,2]. According to thirteen studies from different countries that provided data on therapeutic modalities (Table 2), the rates of patients with acromegaly that have undergone any pituitary-directed surgery at least once as part of their treatment range from 55% to 95% [7, 11–15,17–19,39–41,43–45], and 44% to 90% for TSS. Taiwan and South Korea had highest rates of TSS, the chief treatment strategy for 95% and 90% of patients, respectively. Normally, the goals of acromegaly treatment include biochemical normalization of GH and IGF-1 levels, tumor mass reduction and systemic comorbidity prevention. Owing to different criteria for remission, follow-up period and treatment modalities, it is difficult to review the remission rates among different studies, but generally, overall surgical cure rate was reported as 40% to 70% [1,2]. According to a review by Chen et al. [47], microscopic and endoscopic resection of somatotropinomas are both suitable approaches for TSS, generating remission rates over 60% at final follow-up.
Table 2
| Study | Country | SURG, % | TFS, % | TSS, | U-SURG, % | RT, % | GKS only, % | RT only, % | GKS+ RT, % | Y Imp, % | U-RT, % | MED, % | DA, % | BR, % | CAB, % | SSA, % | OCT, % | LAN, % | GHRI, % | None, % | Known, % | Unknown, % | Total |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Kwon et al. [13]/Park et al. [15] | Korea | 90.40 | NA | 90.40 | NA | 4.80 | 3.90 | 0.80 | 0.10 | NA | NA | 2.50 | 0.60 | 0.60 | 0 | 1.80 | 1.60 | 0.30 | NA | 2.40 | 84.20 | 15.70 | 1,350 |
| Reincke et al. [17] | Germany | 59.80 | 3.60 | 44 | 12.10 | 1.40 | 0.32 | 0.26 | NA | 0.45 | 0.39 | 34.10 | 12.10 | NA | NA | 22 | NA | NA | NA | NA | 95.40 | 4.60 | 1,543 |
| Portocarrero-Ortiz et al. [19] | Mexico | 72 | NA | 72 | NA | 2.40 | NA | NA | NA | NA | NA | 26 | NA | NA | Either | Either | NA | NA | NA | NA | NA | NA | 2,057 |
| Hoskuldsdottir et al. [12] | Iceland | 77a | NA | NA | NA | 33a | 6 | 27 | NA | NA | NA | 55.73a | 23.07 | NA | NA | 5.77 | NA | NA | 19.20 | NA | 100 | 0 | 52 |
| Bex et al. [7] | Bel/Lux | 68a | NA | NA | 34a | 0.72 | 32.80 | NA | 0.48 | NA | 78a | 14.80 | NA | NA | 32.10 | NA | NA | 0.96 | 3 | NA | NA | 418 | |
| Mestron et al. [14] | Spain | 81a | 0.08 | 80.92 | NA | 45a | 0.74 | 41.30 | NA | NA | 0.25 | 64.6a | 20.34 | NA | NA | 44.30 | NA | NA | NA | 0.70 | 97.60 | 2.40 | 1,219 |
| Gruppetta et al. [11] | Malta | 79a | NA | NA | NA | 58a | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | 52 |
| Tjornstrand et al. [18] | Sweden | 55 | NA | NA | NA | <1 | NA | NA | NA | NA | NA | 45 | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | 53 |
| Khamseh et al. [39] | Iran | 61 | NA | NA | NA | 27 | NA | NA | NA | NA | NA | 12 | NA | NA | NA | NA | NA | NA | NA | 0 | NA | NA | 85 |
| Tseng et al. [40] | Taiwan | 95a | NA | 85.10 | 9.90 | 27.9a | 15.40 | 7 | NA | NA | 4.80 | >90a | NA | 22.40 | 34.20 | NA | 60.30 | 30.50 | NA | NA | NA | NA | 272 |
| Maione et al. [43] | France | 80a | NA | NA | NA | 17a | NA | NA | NA | NA | NA | 71a | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | 999 |
| Daly et al. [41] | Belgium | 89a | NA | NA | NA | NA | NA | NA | NA | NA | NA | 44a | NA | NA | NA | 44 | NA | NA | NA | NA | NA | NA | 9 |
| Petrossians et al. [44] | EU Multicenter (10 countries) | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | 3,173 |
| Vallette et al. [45] | Canada | 89a | NA | NA | NA | 22a | NA | NA | NA | NA | NA | 64.5a | NA | NA | 17.10 | 60.90 | NA | NA | 9.90 | NA | NA | NA | 649 |
SURG, any kind of surgical excision of adenoma; TFS, transfrontal surgery; TSS, transsphenoid surgery; U-SURG, unknown surgery type; RT, any kind of radiotherapy; GKS, gamma knife surgery; GKS+RT, combined use of gamma knife surgery and radiotherapy; Y Imp, yttrium implants; U-RT, unknown radiotherapy; MED, any kind of medical treatment; DA, dopamine agonist; BR, bromocriptine; CAB, cabergoline; SSA, somatostatin analogue; OCT, octreotide; LAN, lanreotide; GHRI, growth hormone receptor inhibitor; NA, not available.
![]()
If surgery is contraindicated or if the goals are not achieved, then medical therapy is resorted to, as there is a visible trend of decreasing use of radiotherapy over time, potentially due to extensively delayed remission [2,43]. Multiple studies reported that radiotherapy (conventional and stereotactic radiosurgery) was applied as primary therapy at rates that range between <1% and 27% [13,15,17–19,39]. Stereotactic radiotherapy, specifically gamma knife surgery (GKS), is preferred to conventional radiotherapy, as it normalizes GH and IGF-1 levels more effectively [2] and achieves remission rates of 59% to 65% when used as adjuvant treatment for residual or recurrent tumors [48–50]. Adverse effects of GKS, such as time-dependent hypopituitarism (12.3% to 35% after 5 years), visual disturbance, and brain necrosis have been observed in some patients [48–50].
Pharmacotherapy was prescribed as primary treatment in 2.5% to 45% of patients among the reviewed studies, while the majority was reserved for post-operative adjuvant treatment [13, 15,17–19,39]. To date, SSAs, like octreotide, and lanreotide have been the most commonly used drugs, being used at least once in a given patient in 6% to 61% of the time [7,12–15,17,41, 45]. SSA achieve biochemical remission in approximate 55%, while tumor size shrinkage is observed in 80% of the cases [1,51]. The latter finding makes SSA a suitable pre-operative medication for decreasing perioperative morbidity and tumor volume, as well as improving surgical outcome [52]. The effect on remission rates and recurrence (15% to 84%) after SSA discontinuation is highly variable [53], although complete remission of 4 years after discontinuation was reported in a case [53]. As one of the examples of SSA intolerance, SSA-associated deterioration in glucose metabolism via inhibition insulin secretion has recently been reported, which can be predicted by plasma glucose 120 minutes post-oral glucose tolerance test (OGTT) [35]. Unfortunately, 20% to 25% of acromegaly patients are resistant to SSA, requiring the use of either DAs or growth hormone receptor antagonist (GHRI). To compare the effectiveness of DA and GHRI, further investigation is required about the medical outcome of patients in different countries. Cabergoline is preferred to bromocriptine and is reserved as either primary or secondary treatment for patients with mild disease with close to 35% achieving remission [1,51,54]. Pegvisomant, the representative GHRI, is also mainly indicated for patients with resistance to first-line SSA [1,55].
Combined medical treatment has been on the rise, as reported by Strasburger et al. [56]. SSA and DA combination may return IGF-1 levels to normal in over half of the patients [1,54,57]. SSA and GHRI combination (SSA+GHRI) may lead to 97% and 67.5% normalization in IGF-1 levels in clinical trials and 5-year surveillance studies, respectively [1,54,57]. The latter combination has shown higher rates of liver enzyme elevations, implying the need for caution [54]. Furthermore, DA and GHRI combination (DA+GHRI) in clinical trials has shown more efficacy in combination, than singularly, introducing them as substitute for patients intolerant or fully resistant to SSA [54]. GHRI monotherapy, SSA+GHRI and DA+GHRI have a similar incidence in adverse events, suggesting the high efficacy and safety for pegvisomant use [56]. Further prospective research is required to provide more evidence about the long-term effectiveness of combined approach to medical therapy.
Recently, recommendations have been published about implementing adjunctive chemotherapy, temozolomide, in patients with serious complications from surgery or radiotherapy, or as last-line therapy [54,58]. It is unclear; however, if it is more effective as monotherapy or in combination. Further research is also required in the novel manipulation of chemokine receptor CXC chemokine receptor 4 (CXCR4), as a target for anti-neoplastic agents, like its antagonist, d-Arg3FC131, which has shown to induce apoptosis and inhibit tumor growth in nude mice [59–61]. Moreover, gene therapy, being a large field of opportunities for the exploration of new treatment methods, is also a field of uncertainty in the resultant adverse effects of genetic modification, such as inserting SSAs into viral vectors to restrict its expression to the pituitary [62].
Go to :
PRECISION THERAPY & PREDICTORS OF RESPONSIVENESS
“One treatment for all” concept cannot be applied for the significant proportion of patients with persistent disease after surgery or resistance to medical therapy. Initial selection of SSA and subsequent switch to another drug type, judging by the absence of disease control, have most perilous disadvantages, when considering the delay in cure and worsening of patient quality of life [22]. Individualized management guidelines [20] can be directed by the advances in translational research, which can yield markers of responsiveness to surgery or medical therapy.
Clinical markers include gender [63,64], age [24,63,64], initial post-operative GH and IGF-1 levels [64], tumor invasion [2,64], tumor size [9,24], surgical method [55,63,65,66], surgeon experience [63,66], and intensity of T2-weighted magnetic resonance imaging (MRI) [23]. Generally, pre-menopausal women exhibited poor surgical remission, having more aggressive tumors with higher cavernous sinus invasion rates, lower post-operative 75 g OGTT GH normalization and lower long-term surgical remission rates [63,64]. Old age was associated with microadenomas, based on the hypothesis of gradual biological decline in GH and IGF-1 levels [24]. In South Korea, overall surgical remission rates were above 85% in intrasellar adenomas and 50% to 70% in extrasellar adenoma [2]. Aggressive surgical resection should be applied in tumors with cavernous sinus invasion that is confined to the medial compartment of the internal carotid artery and even in surgically incurable tumors, which can aid remission by minimizing tumor burden and aiding effects of adjuvant therapy [66]. Another study also showed that it does not increase the risk of postoperative hypopituitarism [63]. Removal of tumor pseudocapsule during transsphenoidal adenomectomy (TSA) improves endocrinological remission rates and no increased risk of postoperative hypopituitarism or cerebrospinal fluid rhinorrhea [55,65]. Somatotropinomas present as macroadenomas (>10 mm in diameter) in 65% to 89% (Table 1) [7–21,39–46], decreasing the possibility of complete surgical cure and evoking persistent disease in >60% of cases [9]. Gamma knife surgery should be avoided in patients with cavernous sinus invasion, owing to the risk of hypopituitarism and radiation necrosis [49]. Moreover, somatotropinoma T2 signal hypointensity on MRI was related to better clinical outcomes with SSA use [23].
Long-term remission and the selection of the type of adjuvant therapy was assessed using the predictive role of immediate and 1-week post-operative nadir GH levels <1.0 mg/L [67]. A criteria for biochemical remission after surgery of <0.4 mg/L nadir GH concentration with 75 g OGTT [68] and a treatment goal of <1.0 mg/L [69] random GH concentration have been proposed. However, it was shown that neither cut-off values were superior in predicting better improvement of metabolic levels in acromegalic patients [70].
Patients with acromegaly cured with TSA may experience GH deficiency (GHD), which may lead to cardiovascular, bone and psychological complications. Ku et al. [71] found that the presence of a bilateral pituitary tumor and low immediate post-operative 72-hour GH concentrations (below 0.23 mg/L) predicted GHD. Administration of recombinant GH has been shown to improve IGF-1 concentrations and quality of life without deleterious adverse effects [72,73]. Tritos et al. [74], however, warned of increased cardiovascular complications in GH replacement and recommended further investigation on whether it was due to previous GH excess in acromegaly.
Histological markers of remission or resistance to medical treatment comprise of granulation pattern [56], expression of somatostatin receptor subtype 2a (SSTR2a) [25,26,28], SSTR5 and dopamine D2 receptor (DR2) [34], Ki-67 index [30], β-arrestin, E-cadherin and Raf kinase inhibitor protein (RKIP) [31–33]. Tumors with sparse granulation pattern, with hypointensity on T2-weighted imaging [23] and low SSTR2a expression [26], exhibited more invasion and little to none GH response [26]. SSTR2 and 5 are the receptors on which SSA exert their effects, but only the expression of the former has been linked to positive predictive response to treatment [25,28]. High SSTR2a expression is a predictor for short-term and long-term SSA responsiveness by normalizing biochemical levels and reducing tumor size [25,28]. Depletion of SSTR2 expression was recognized in SSA pre-treatment groups, showing the loss of sensitivity with sustained SSA use [25,28]. Increased DR2 expression, as the principal receptor subtype expressed in somatotropinomas, exhibits better response to DA, while increased DR1 or DR5 expression may be related to resistance [34]. Ki-67 index >3% on histopathological analysis of GH-secreting tumors is associated with increased resistance to treatment [30]. Low expression of β-arrestin, E-cadherin, and RKIP have the potential to be positive predictors of responsiveness, but further research is required to finalize current results [31–33].
Molecular markers of long-term remission or cure consist of aryl hydrocarbon receptor-interacting protein (AIP) mutation and pleomorphic adenoma gene 1 (PLAG1) like zinc finger 1 (ZAC1) expression [27,29,30], guanine nucleotide binding protein, alpha stimulating 1 (GNAS1) [29], somatostatin receptor subtype 5 (SSTR5) variants, SST5TMD4 and SST5TMD5 and single nucleotide polymorphisms, C1004T and T-461C mutations [36–38]. Germline inactivating mutations of AIP, causing increased ZAC1 protein expression, are present in 30% of familial isolated pituitary adenomas cases of acromegaly [29,30]. In addition to causing predisposition to more aggressive and larger pituitary neoplasms, they tend to cause disease in younger patients [29]. Pioneering an animal model for somatotropinomas, namely somatotroph specific AIP knock out (sAIPKO) mice, allowed for the study of familial acromegaly and adult-onset GHD [27]. Activating mutations of GNAS1, encoding the stimulatory alpha-subunit of protein G, have also been associated with excess GH secretion in 20% to 30% of McCune-Albright Syndrome patients [29]. The application of the two truncated isoforms of SSTR5, SST5TMD4 and 5, and two of its alleles, C1004T and T-461C, in a clinical setting is yet to be finalized [36–38]. Another potential predictor of responsiveness to treatment could be the increased expression of specific circulating microRNA (miRNA), such as miR-338–3p, found to act as a key promoter of proliferation, migration and invasion in somatotropinomas [61].
Go to :
CONCLUSIONS
Nowadays, personalizing management of acromegaly does not persist in theory, but is slowly being integrated into reality. One of the routes to nearing precision therapy is expanding epidemiological databases: adding much needed geographical and ethnic comparisons of patients and sharing treatment approaches, especially in Asia. The solidified incidence rate of 3.6 to 3.9 cpm/year in South Korea [13,15] is not only similar to current Western literature [7–12,14,16–19,39–45], but also can be a standard for epidemiological comparisons of acromegaly among other Asian countries. Another route of achieving customization of treatment: begin implementing specific predictors of surgery and adjuvant treatment responsiveness. Clinical [2,24,55,63–66] and imaging [9,23,24] features of the patient, histological and molecular characteristics of the tumor [23,25,26,28–30,34] (more details in Fig. 1) may be coalesced into clinical guidelines [20], while more markers are being researched [29,31–33,36–38,61].
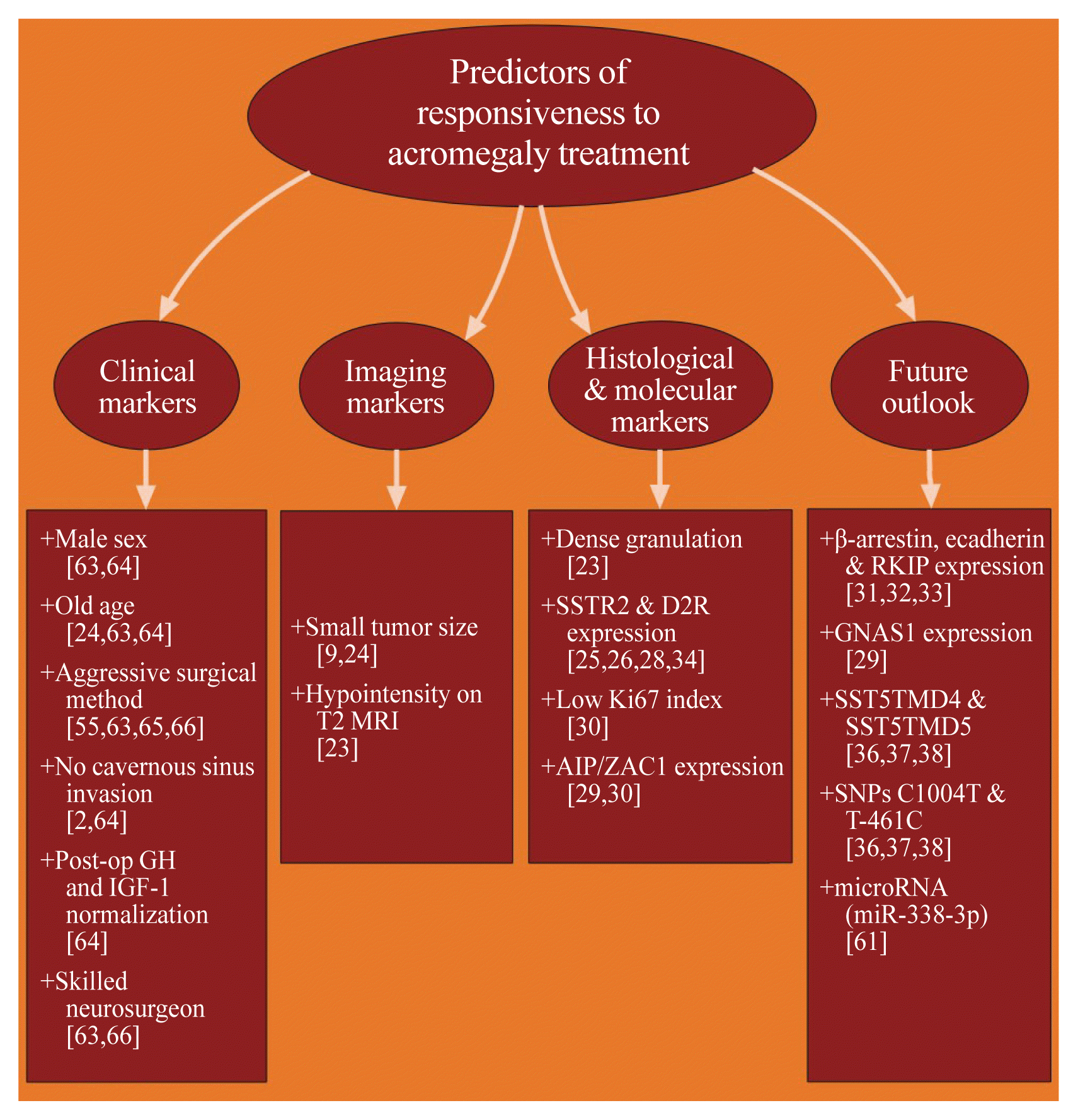 | Fig. 1Flowchart representing current clinical, imaging, histological and molecular predictors, as well as future predictors whose role can be finalized with developments in translational research [2,9,23–26,28–38,55,61,63–66]. GH, growth hormone; IGF-1, insulin-like growth factor-1; MRI, magnetic resonance imaging; SSTR2, somatostatin receptor subtype 2; D2R, dopamine d2 receptor; AIP, aryl hydrocarbon receptor interacting protein; ZAC1, pleomorphic adenoma gene 1 like zinc finger 1; RKIP, Raf kinase inhibitor protein; GNAS1, guanine nucleotide binding protein, alpha stimulating 1; SST5TMD4, somatostatin receptor subtype 5 splicing variant; SNP, single nucleotide polymorphism.
|
Go to :
 XML Download
XML Download