

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Acute myeloblastic leukemia (AML) is a hematologic disorder, which is due to accumulation of myeloblasts in the bone marrow [12]. Chemotherapy is the prevalent standard treatment for the AML. 60–80% of cases achieve complete remission, but 50–70% of them experience a relapse [3]. The AML relapse is attributed to leukemic stem cells (LSCs), which are capable of re-initiating the malignancy and are assumed to be rescued from chemotherapy [4]. Allogeneic hematopoietic stem cell transplantation (allo-HSCT) is the most commonly chosen treatment approach in AML patients. Donor lymphocyte infusion (DLI) is considered as another treatment for AML. Graft-versus-host disease (GvHD) is one of the reasons of urgent requirement for using novel and effective treatment options in AML [5]. One of the main reasons to find and use new therapeutic methods for the treatment of AML is to root out LSCs. Also, these strategies are favorable since they lead to smaller number of complications and target the bulk of blasts compared to chemotherapy [1]. For this purpose, much attention has been paid to the identification of the cell marker targets on the LSC cell surface [6]. Among the cell surface markers, CD33 antigens are highly promising candidates for AML targeted therapy. The high expression level of CD33 in AML blast had been already shown and reported [78]. CD33 was detected on blasts of most AML patients as well as on normal myelocytes [8]. Ideally, it seems that CD33 surface antigens are limited to LSCs [9], but absent on normal HSCs, making them an interesting target for AML therapy [1011]. On the other hand, CD marker-specific chimeric antigen receptor (CAR)-T cell therapies in acute lymphoblastic leukemia (ALL), as well as antibody–drug conjugates, have been investigated to develop cellular immunotherapy-based AML. It has been shown that development of cancer stem cell (CSCs)-selective therapies is important for treating cancer [12]. This review discusses CAR-T cell approaches as well as monoclonal antibody (mAB)-based therapy, the two antibody-based therapies for the treatment of AML.
CANCER STEM CELLS
Recent documents have indicated that CSCs are a subpopulation of stem-like cells within tumors that are capable of differentiation, tumorigenicity, and self-renewal. In addition, these cells express a set of cell surface markers such as CD133, CD44, CD33, CD29, CD24, aldehyde dehydrogenase1 (ALDH1), and others, which can be enriched by other cells within the tumor [13]. The first documented role of CSCs was published in a study by Lapidot et al. [14] (1994) in which an AML-initiating cell (CD34+/CD38−) was identified in human AML by transplanting into immune deficient mice. The subsequent reports identified human CSCs in solid tumors of the breast, brain, and colon [1516]. Of particular interest, the expression of CSC markers is tissue- and tumor type-specific. For example, CD34+CD8− was characterized for leukemia, CD44+CD24− for breast, CD44+ for head and neck, and CD133+ for lung, brain, colon, and others [17]. In regard to the origin of CSCs, there are two prevailing theories. The first theory proposes that CSCs arise from normal progenitor cells that have the ability to generate tumors when they encounter environmental alterations. The second theory posits that CSCs arise from normal somatic cells that acquire malignant behavior through genetic mutations [18].
LEUKEMIC STEM CELLS (LSCS)
Since LSCs were first found in AML in 1997, a wide range of studies have been conducted to characterize and identify populations of different cells in various tissues [19]. LSCs are typically considered to be populations of cells that are capable of proliferation, differentiation, and self-renewal, and that play a significant role in the occurrence of leukemia [20]. Generally, LSCs do not have to arise from a normal stem cell, and the cell origin for most LSCs has not yet been determined [21]. The first theory, mentioned above, is that LSCs are able to arise from normal HSCs, and the other indicates that genetic mutation in HSCs causes them to become LSCs [21]. It has been indicated that LSCs are regulated by signaling pathways and particular surface antigens [22]. For the first time, Bonnet and Dick [23] (1997) described that only leukemic cells, which express similar markers as normal hematopoietic stem cells (CD34+ CD38−), have the ability to initiate hematopoietic malignancy, and they named these cells LSCs. Studies have indicated that AML LSC markers are considerably dissimilar. AML LSCs specifically possess CD34+CD38− cell surface markers [19]. CD123 and CD33 markers are extremely promising candidates for AML targeted therapy among the abovementioned cell surface markers [6].
HETEROGENEITY OF LSCS IN HUMAN AML
During the past few decades, an increasing number of studies have demonstrated that AML may occur predominantly in either multipotent HSCs or mature committed myeloid progenitors [24]. In some leukemias, experimental studies have reported the considerable influence of the clonal process in numerous cell lineages (monocytes, erythrocytes, and occasionally B-lymphocytes), which reflect the source and expansion of AML at the level of pluripotent stem cells [24]. In other leukemias, it was proposed that CD33− precursors would be significantly or entirely normal. Bernstein et al. [25] (1992) eliminated CD33− cells in vitro through FACS in a few patients with leukemia in order to investigate this hypothesis, and they placed the remaining CD33− cells in long-term culture along with the irradiated allogeneic stroma cells. By passing the time, in some patients, the colony-forming cells (CFCs) with X chromosome inactivation models are consistent with non-clonal hematopoiesis significantly which were really generated by CD33− precursors [25]. Moreover, immunophenotypic variants have been distinguished by some studies using differentiation markers that differed between normal LSCs and HSCs [26], and later xenotransplantation studies demonstrated that the transformation process may happen in progenitor cells during the stem cell stage [27]. In the previous cited studies, AML was reconstituted in immune-deficient mice from cells that phenotypically seemed to be more mature compared to pluripotent HSCs [27]. Moreover, in one later study, it was proposed that leukemic cells, which engraft in immune-deficient mice, are phenotypically more close than HSCs, the LSC-containing cell fraction [27]. Also, a new finding has shown the feasibility of engrafting CD33+-derived cord blood cells with multi-lineage hematopoiesis [28].
CELL-BASED THERAPY IN AML BASED ON STEM CELL HETEROGENEITY
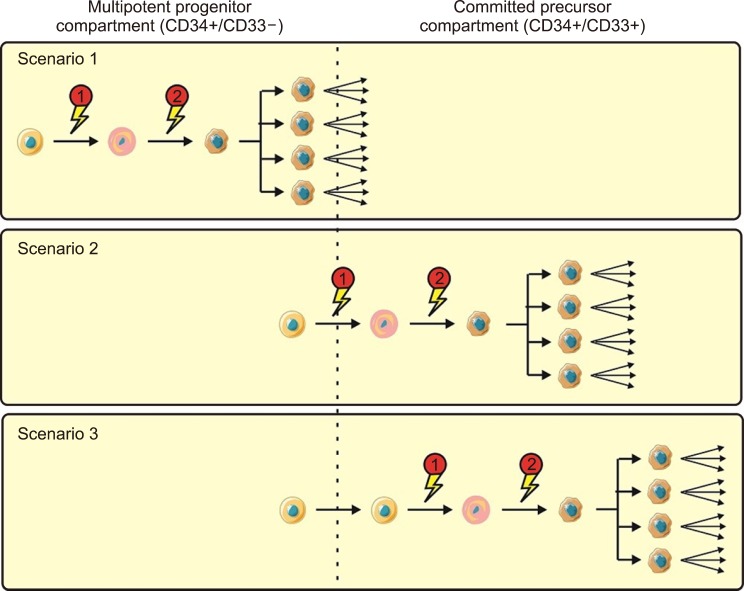
Though against restrictions, Recent investigations have proposed three possible testable scenarios of AML development (Fig. 1) [22829]. In scenario 1, both the preliminary and subsequent mutations occur at the level of pluripotent CD33− progenitors; these are referred to as “immature” leukemias. Here, clonal dominance is limited to monocytes and granulocytes or is developed in multiple cell lineages [30]. Some of immature leukemias may be recognizable in xenotransplantation examinations by the capability of CD34+/CD38− cells to recapitulate the disease [2]. On the contrary, only one mutation happens at the committed myeloid cell level in “mature” leukemias. The preliminary mutation specifically occurs in pluripotent HSCs in the second scenario; however, the cooperating mutations result in the full transformation to AML, and then clonal expansion only happens at a later stage, perhaps at the committed CD33-myeloid progenitor level [2]. Eventually, both of mutations and clonal expansion happen at the level of committed CD33-myeloid progenitors in the third scenario [31]. Acute promyelocytic leukemia (APL) may be an example of the third scenario. It is possible that the character of LSCs has considerable prognostic notions.
CD33 ANTIBODY BASED THERAPEUTIC APPROACHES IN AML
The myeloid differentiation cell surface marker CD33 has been the target in antibody-based therapeutic approaches for AML. Immuno-targeting methods are designed to target specified antigens that are expressed on the leukemic cell surface, and these methods are another extensively discussed new approaches for AML described below [32]. For a few decades, a preliminary focus of antibody-based therapy in AML has been CD33. CD33 is a membrane-bound protein of the Siglec family that is expressed by HSCs. CD33 is a critical molecule in the inflammatory response and is found to be expressed in up to 90% of AML blasts [33]. Clinical investigations have shown that gemtuzumab, the humanized anti-CD33 antibody, binds to CD33-antigens and enters the cells through endocytosis [34]. The low expression and slow entrance of CD33 complexes lead to comparatively limited CD33-mediated drug uptake per unit of time [35]. Various novel CD33-targeted therapeutics, which may overcome the limitations of earlier therapeutics, are currently under consideration in preclinical studies [3637]. Primitive efforts at targeting CD33 with unconjugated antibodies were discouraging [38]; however, the CD33 receptor's endocytic properties make it well-suited for antibody–drug conjugate (ADC)-based therapies. ADCs are known as engineered molecules of monoclonal Ab conjugated to a potent cytotoxic agent through stable linkage that are targeted for tumor-associated antigens [39]. “Mature” leukemias may benefit individually from stem cell-based therapies because their underlying LSCs may be different from normal LSCs. Then, LSC-targeted therapy could be partially substituted by other therapeutics. As mentioned earlier, we focused on CD33 as the myeloid differentiation antigen, which is predominantly expressed on leukemic blasts in most AML patients (85% to 90%) [35]. The practicality of CD33-targeted therapeutics is supported by the observation that, in some leukemias with clonal influence limited to monocytes/granulocytes, ablation of CD33+ cells could result in the resumption of normal hematopoiesis [33]. In other words, unconjugated anti-CD33 antibodies were greatly useless in patients with distinct CD33+ non-APL AML [40]. mAb-based therapy for AML includes gemtuzumab ozogamicin (GO), an anti-CD33 mAb that is conjugated to calicheamicin, an antitumor antibiotic, and lintuzumab, an unconjugated anti-CD33 mAb. Taken together, it is clear that mAb-based therapies are an integral part of the therapeutic tactics for leukemia, though they have been less favored in AML than in other hematologic disorders [38]. Using CAR-T cell therapy against AML cells is another alternative method to treat AML patients [41].
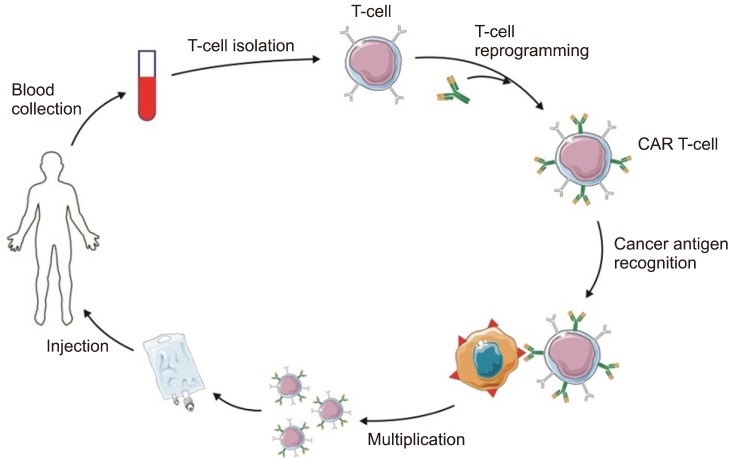
CAR-T CELL THERAPY
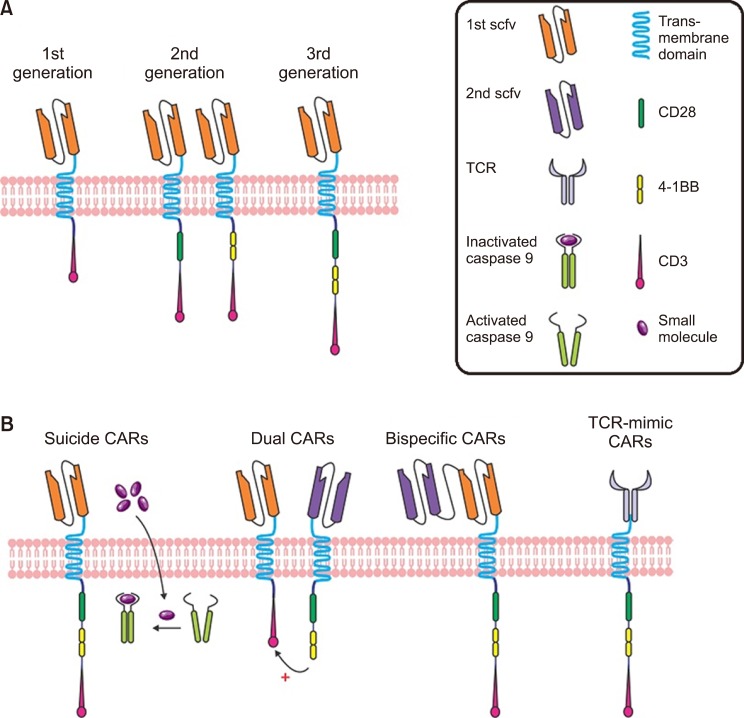
The notion of optional T cell therapy was preliminarily drawn from the discovery of the proposed cytotoxic T cells as the main players in mediating allo-HSCT [42]. Subsequently, engineered cytotoxic T cells were tested in clinical trials. CAR-T cells and T cell receptor (TCR) engineered T cells are considered among the most important technologies that utilize genetically engineered cytotoxic T cells. These two methods immediately place the T cell in proximity to the antigen-bearing target cell. Accordingly, one main difference between these and earlier therapies is that the extracellularly and intracellularly expressed antigen receptors (especially HLA) are identified by a TCR; however, the CAR-T cells are HLA-self-determining, and recognize only antibody-specific surface antigens. Fig. 2 shows the constituent components of each [5]. There have been three generations of CAR-T cells. The first-generation contains only the tyrosine-based zeta-signal-transducing subunit from the TCR/CD3 receptor complex [43]. Near this zeta-domain, in the later generations, extra costimulatory molecules such as CD27, CD28, CD134, CD137, etc. are anchored: only one is anchored in the second generation, and while there are two in the third generation [4445]. When regression happens after CAR-T cell therapy, cancerous cells generally miss the targeted antigen. This is resolved by CAR-T cells that target multiple antigens [46]. These CAR-T cells are named dual-targeting T cells [4748]. Up to now, only a few CAR-T cells have been considered in clinical trials for AML, and unlike B-cell disorders, no licensing authority has approved CAR-T cell therapy for AML. Hofmann et al. [5] (2019) have assembled an overview of antigen which are possibly appropriate for CAR-T cell therapy in AML, as well as those that are previously considered in clinical trial.
CAR-T cell therapy would require an antigen that is particularly or specifically expressed on AML cells [49]. In the absence of some AML-specific target antigens, this immunotherapy reaches their toxicity approach quickly and can no longer be used because of the on-target killing of normal non-AML myeloid cells which are fundamental for maintenance and survival [50]. In other words, a major challenge for CAR-T cell therapy of AML patients is to find leukemia-specific target antigens [51]. As mentioned above, CD33 is a noteworthy target for immune-cellular therapy against AML. This is represented by the expansion of gemtuzumab, a drug-conjugated anti-CD33-antibody. However, while it was first approved in 2000 by the FDA, it was retracted from the market in 2010 due to bone marrow toxicity. It was reintroduced in 2018 after a meta-analysis by Hills et al. [52] (2014). Their report indicated that a fractionated, low dose of gemtuzumab in combination with chemotherapy led to ameliorated overall maintenance of 280 treated AML patients [52]. Up to now, one report of a patient with r/r AML, who was treated with anti-CD33 CAR-T (CART-33) cell, has been published [53]. In that study, a phase 1 clinical trial was conducted to evaluate the feasibility and efficacy of CART-33 cells for the treatment of advanced AML patients. It was described that the injection of CART-33 cells alone could result in degradation of disease at the early stage, which indicates the strong in vivo cytotoxic effect of CART-33 on CD33+ blasts. Nevertheless, CD33+ leukemic cells were gradually eliminated until a florid advancement was observed in the later stages of cell therapy. The high level of steady data from CART-33 molecules in vivo and the survival of the cytotoxic activity of lymphocytes isolated from the patients' peripheral blood on CD33+ HL-60 illustrated a feasible mechanism for CD33-directed therapy against leukemic cells with low CD33 expression. Moreover, mentioned toxicities which are related to the tissue dispensation of the targeted antigen, a cytokine release syndrome or inflammatory response syndrome has been frequently stated with CAR-T cell infusions and its intensity is extremely accompanied by tumor lysis [5354].
CONCLUSION
There is an increasing number of targeted therapeutic tactics to treat AML; nevertheless patient outcomes have remained poor, and clinicians have an urgent need to find new treatment strategies for patients. This is probably because of the heterogeneous and extremely polyclonal character of AML, and approaches that target peculiar mutations in AML cells may result in the eradication just single subclasses, and thus they are insufficient to rid a majority of patients of the disease. As it was discussed in this work, CAR-T cells are starting to build a new therapeutic category in clinical practice, but the extent of CAR-T cell immune-therapy for AML still needs to be investigated. There are important hurdles to surmount: determining the appropriate antigen with low “off-tumor” toxicity, for example, and also discovering methods to minimize the “off-tumor” toxicity. The CAR competition has been started, and hopefully, it will ameliorate AML and fortify the therapeutic armentarium against the disease (Fig. 3).
 XML Download
XML Download