

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Dear Editor,
Congenital stationary night blindness (CSNB) is a non-progressive, clinically and genetically heterogenous group of retinal diseases characterized by various clinical features, such as night blindness, visual decrement, myopia, nystagmus, and/or fundus abnormalities. Seventeen gene mutations have so far been identified to be associated with CSNB and are related to phototransduction cascades, photoreceptor to bipolar cell signal transmissions, and/or retinoid recycling in the retinal pigment epithelia [12]. Among those 17 genes, TRPM1 encodes the transient receptor potential cation channel, subfamily M, member 1 (TRPM1) located in the ON-bipolar cell, and gene mutation leads to loss of the ON-bipolar response, resulting in CSNB [3]. Although a few cases of CSNB have been reported in Korea, all of the patients were diagnosed based on fundus examination and standard electroretinogram (ERG) without genetic analysis. Therefore, for the first time, we report a case of CSNB in a Korean patient with pathogenic novel mutations in the TRPM1 gene detected via genetic analyses.
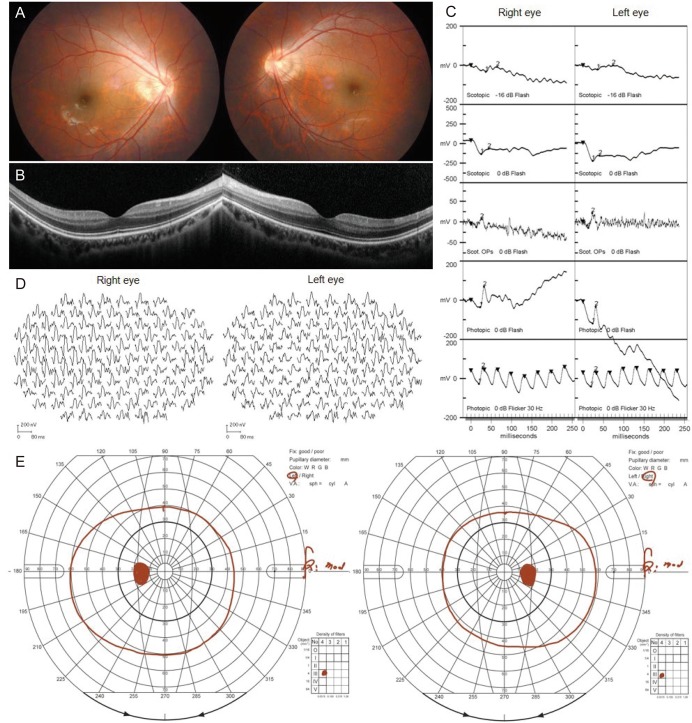
A 19-year-old male visited our ophthalmology clinic with symptoms of night blindness. He had undergone three strabismus surgeries for exotropia. His corrected visual acuity was 20 / 50 in the right eye and 20 / 30 in the left eye, and his cycloplegic refractive error was −6.50 Dsph = −0.50 Dcyl × axis 45 in the right eye and −6.50 Dsph in the left eye. Dilated fundus examination showed no abnormalities (Fig. 1A). Optical coherence tomography showed normal macular structure in both eyes (Fig. 1B). However, standard ERG showed markedly decreased b-wave and oscillatory potential, with a negative ERG and a normal cone response in both eyes (Fig. 1C). Multifocal ERG (Fig. 1D) and Goldmann perimetry (Fig. 1E) were normal. Under the clinical diagnosis of CSNB, we performed genetic analysis using a next-generation sequencing-based panel targeting 204 known genes of inherited retinal diseases [4]. In this analysis, we found two novel null pathogenic variants of TRPM1 (NM_001252020.1, c.[3397C>T](;)[3911delA], p.[Arg1133Ter](;)[Asn1304Ilefs]) according to the American College of Medical Genetics and Genomics criteria [4]. Finally, he was diagnosed with autosomal recessive (AR) CSNB with TRPM1 mutations. He showed no disease progression throughout two years of follow-up.
CSNB can be classically divided into four types according to ERG pattern and fundus appearance: Riggs type, Shubert-Bornschein type, fundus albipunctatus, and Oguchi disease. The Riggs type is characterized by scotopic a-wave reductions on ERG with normal fundus appearance. The Shubert-Bornschein type is identified by electronegative ERG with normal fundus appearance and is divided into two forms: complete-type CSNB (cCSNB) and incomplete-type CSNB, respectively characterized by ON- or both ON- and OFF-bipolar pathway dysfunction. Fundus albipuntatus is noted by recovery of scotopic responses after prolonged dark adaptation on ERG and small white or yellow flecks throughout the retina. Lastly, Oguchi disease is characterized by delayed rod-dark adaptation on ERG that fully recovers with disappearance of golden-yellow discoloration of the retina. Also, the inheritance patterns of CSNB are X-linked (XL), AR, or autosomal dominant [2]. In our case, the patient had AR cCSNB of Shubert-Bornschein type. With advancement in molecular analysis techniques, 17 gene mutations in CSNB patients have been identified [2], as mentioned above. For example, in cCSNB, mutations of genes such as NYX with XL inheritance pattern and GRM6, TRPM1, GPR179, and LRIT3 with AR pattern have been found. Furthermore, CACNA1F mutation with XL inheritance pattern and CABP4 and CACNA2D4 mutations with AR inheritance pattern have been found in incomplete-type CSNB [25]. It is likely that additional associated genes will be identified in the future.
The limitation of our report is that we could not verify whether the two variants of TRPM1 were compound heterozygotes with in trans position since genetic analysis of the parents was not feasible. Considering the predicted pathogenicity of the two variants, however, both can be assumed to be involved in the pathogenesis of AR CSNB in our patient.
In conclusion, we report a case of AR CSNB with novel TRPM1 gene mutations in a Korean patient. This report may help improve the understanding of the pathophysiology and treatment in CSNB patients.
 XML Download
XML Download