

 PDF
PDF Citation
Citation Print
Print




INTRODUCTION
Human leukocyte antigen (HLA) plays an important role in regulating the immune response.1 In particular, the self-recognition function is essential when transplanting donor organs that are not consistent with HLA typing in recipients, as it is a major cause of severe transplant rejection. Therefore, transplant recipients should be tested for HLA typing before transplantation to confirm a compatible HLA type with the donor.2 Moreover, HLA diversity was recently reported to be associated with more than 100 diseases as well as severe drug hypersensitivity.345 However, the HLA results of transplant patients and donors are rarely used for the diagnosis of HLA-related diseases or for predicting future adverse drug reactions. One of the reasons for this underuse of HLA typing results in clinical settings outside of the context of organ transplantation is the variable level of resolution depending on the testing methods, ranging from a micro-lymphocytotoxicity-based assay to next-generation sequencing.6 Developments in next-generation-sequencing technology have now enabled the high-resolution screening for alleles and genotypes as well as serologic typing.7 Even in the case of organ transplantation, the resolution of the tests required differs depending on whether the target organ is a solid tissue or blood. However, another, and arguably more important, reason for the underuse of HLA typing is the lack of standardization and structure in the representation format of the HLA typing results in electronic medical records (EMRs).
Standardization with respect to the terminology and format of clinical data has been increasingly emphasized owing to advantages of the continuous utilization of essential clinical information from the EMR while minimizing any loss of data during transformation.3 Because the clinical documents written in a free-texted or semi-structured format frequently contain abundant and/or substantial clinical information, it is vital to transform such records to a structured and standardized format, especially when designing a clinical decision support system (CDSS) to ensure patient safety.8 One of the most widely used methods to extract precise data from a free-texted clinical document is natural language processing (NLP)910 and many tools have been developed for retrieving various types of data from EMR using NLP to enable its secondary use.910111213 In particular, genomics applications have been actively studied for extracting phenotype information using NLP from EMR.1415 However, for the extracting genotype data using these NLP-based methods, most studies have thus far focused on literature databases rather than the EMR itself. In other words, little research has been done on the extraction of genotypes stored in EMR for secondary use.
To overcome these limitations and promote the secondary use of essential and available clinical information, in this study, we evaluated the accuracy of extraction methods which focused on the reports of HLA tests obtained by various typing methods with various degrees of resolution that are stored in a semi-structured format at the clinical data warehouse (CDW) of Seoul National University Hospital (SNUH). We further developed a set of rules for extracting and transforming these data into a structured, standard format. The established rules were evaluated by comparison of a validation set generated by manual curation from the testing set. The findings of this study can expanded the possibility of the secondary use of currently underused genotype data for diverse clinical and medical purposes.
Go to : 
METHODS
Data source
We used the data stored at the CDW of SNUH, SUPREME®, to retrieve the HLA reports. The test names used to query of the HLA reports from SUPREME were “HLA-[A,B,Cw,DR,DQ] (DNA,[Low,High])”, in which the terms in parentheses were iteratively used. To develop rules and codes for extracting HLA serotype/genotype data from the free-texted HLA reports, we queried HLA typing results obtained between January 1, 2000, and June 30, 2018 at SNUH as a rule-development dataset. To validate and evaluate the performance of the rules and codes developed using the rule-development set, we generated a second HLA data set by querying reports generated from July 1, 2018 to June 30, 2019 as a test set.
The HLA reports were retrieved in Excel files for each rule-development and test set. In both the EMR and CDW, a single HLA report itself was stored in free-texted format and extracted as a single cell of an Excel file for each patient. As shown in Fig. 1, an HLA report resembled the format of a table with spaces and line breaks to express the structure of attribute-value pairs of HLA serotypes, but was not an actual table.
 | Fig. 1Representative example of raw data in an HLA report. A real example of an HLA typing report with de-identified patient names is shown. An HLA typing result is represented in one cell in an Excel file. This example includes HLA typing results for three patients because most HLA test subjects were recipients of a transplantation procedure, and the physicians wanted to compare all HLA tests of candidate donors with those of a given recipient on the same page on the electronic medical record. In this cell, the HLA test results were arranged in a tabular form using the space bar and a carriage return but were not structured as actual tables with distinct rows and columns. To improve accuracy, we only focused on reports with the HLA typing results of one patient.HLA = human leukocyte antigen.
|
Data extraction method
The extraction process consisted of two steps: 1) develop and apply rules to include reports with only single patient records, and 2) develop and apply rules to extract clinical characteristics and HLA genotypes. We first checked sample HLA reports to determine the pattern of HLA genotypes for developing extraction rules. The iteration processes were applied for 100 reports initially, reaching up to 1,000, and were then applied to the total test set. Based on the first assessment, we developed basic rules and updated codes and then ran the codes to identify missing patterns iteratively. Rule #1 was first used to identify whether or not there was single patient's record in a report. After excluding the reports with the results from multiple patients, we applied Rule #2 to extract clinical data (patient's name and indication of HLA testing) and HLA genotypes for the five main HLA genes (A, B, Cw, DR, and DQ). Because it was relatively simple to establish rules for identifying the number of patients or to extract clinical data in the reports, we mainly focused on extracting HLA genotypes accurately in this step. When application of the rules missed some of the genotypes in the report, we printed them on the monitor to visualize the missing pattern and then developed an additional rule or modified existing rules to extract the missed genotypes. This process was performed iteratively until the remaining missing patterns were only simple spelling errors. For example, there was one report with HLA-B genotyping information only; however, this was not extracted correctly because the genotype was recorded as “HLA-B[multiple spaces][line break][multiple spaces]B5–.” This extraction process was completed using the regular expression function of Python programming language (version 3; Python Software Foundation; https://www.python.org/).
Performance evaluation
The performance of the developed extraction rules was compared with the validation set which was constructed by manual curation (performed by the research nurse and one physician) for the test set. As shown in Fig. 2, after building the primary validation set by the first curator, one of the authors reviewed the primary validation set and compared the result with raw data. After completion of the serial curation process, the final validation set was constructed.
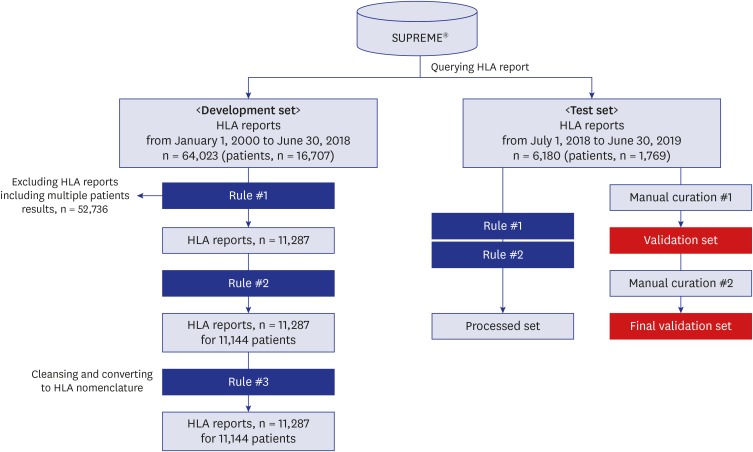 | Fig. 2HLA typing and clinical characteristics extraction pipeline. Rule #1 was designed to exclude HLA reports with typing results for multiple patients. After excluding these reports, we applied Rule #2 to extract clinical variables such as name, sex, and indications of HLA typing. Rule #1 and Rule #2 are extraction rules, and Rule #3 is cleaning rule. Rule #3 was designed to clean the HLA typing results and transform the results to a standard nomenclature. To evaluate the accuracy of the two extraction rules, we applied Rule #1 and Rule #2 to the testing set. The rule-based extraction results of the testing set were then compared with the manually curated results of the testing test, as the validation set. The manual curation process was done sequentially by two different investigators.HLA = human leukocyte antigen.
|
Cleaning and converting the HLA typing to nomenclature
In addition to the extracting rules, we developed rules to clean and convert the extracted HLA typing data to a standard HLA nomenclature format as Rule #3. The WHO Nomenclature Committee for Factors of the HLA System is responsible for determining nomenclature to standardize the expression of these various HLA test results.1617 Because the data used in this study included a mixture of low-resolution serotyping results and high-resolution allele-typing results, we extracted the allele notations according to the HLA subtypes as well as those based on serological specifications. The cleaning and transforming rules were established after extraction of all HLA alleles.
Ethics statement
The study was approved by the Institutional Review Board (IRB) of SNUH (No. 1811-157-989). The IRB approved the conduction of this study without the informed consent from the participants because this study used anonymized retrospective EMR data.
Go to :
RESULTS
Summary of HLA typing data
There were 64,023 HLA reports assigned to 16,707 patients in the rule-development set, and there were 6,180 reports for 1,769 patients in the testing set. There were multiple reports for some patients owing to cases in which physicians ordered tests for HLA genes separately for a given patient or when a patient had multiple potential donors. According to Rule #1, 52,736 reports and 5,420 patients were excluded in rule-development set. Most of the excluded reports were empty because when tests for multiple HLA genes were ordered for a single patient at the same time, the results recorded in a single report were provided in a combined form. There were 4,039 male and 7,105 female patients, including 2,642 high-resolution tests, 5,835 low-resolution tests, and 2,810 tests with undefined resolution in the final rule-development set.
HLA data extraction rules
There were three types of irregularities detected in the HLA typing results from the raw HLA reports. First, the number of patients with test results was inconsistent. In many cases, only one patient's HLA test results was described in a report, whereas other reports included results for more than two patients'. Second, the resolution of HLA typing methods varied, which also influenced the results. Given the rapid development in HLA typing methods, physicians frequently ordered multiple HLA typing methods with different degrees of resolution at the same time for the same patients. Thus, HLA reports based on low-resolution tests, including serology or analysis of sequence-specific oligonucleotides, only provide information on serotypes (allele groups), whereas the reports based on high-resolution methods, including sequence-based typing, provide information on the specific HLA protein or genotype (DNA substitutions). Third, there was ambiguity in the presentation of the HLA typing result. Because there are two allele types for each HLA locus, if the alleles are homozygous, special characters such as quotes were used to express the second allele. In addition, the use of spaces, line separations, and positions of the line break to obtain a table-like format were also irregular. To minimize problem complexity for standardization, in this study, we focused only on the HLA reports with a single patient result according to Rule #1 as described above.
After excluding the reports with the results of multiple patients, we applied Rule #2, which was developed to extract clinical data (patient's name and indication of HLA testing), with a focus on a accurate determination of HLA genotypes as shown in Fig. 2. Through the iteratively developing the rule generation process, including handling missing patterns, we ultimately developed 124 rules to extract HLA genotypes. The patterns of the rules are represented in Table 1 and the specific expressions used for representation are available at https://github.com/geffa/HLAgenotypeParsing/blob/master/RegularExpression. The summary of the initial extraction of the HLA genotypes and clinical characteristics for the rule-development set is provided in Table 2. The extraction results demonstrated an uneven distribution in the number of patients that received typing for each of the five HLA gens. Specifically, HLA-B was tested most frequently (ordered for 69.7% of patients in the rule-development set) and the typing of HLA-C was the least frequent (10.4%).

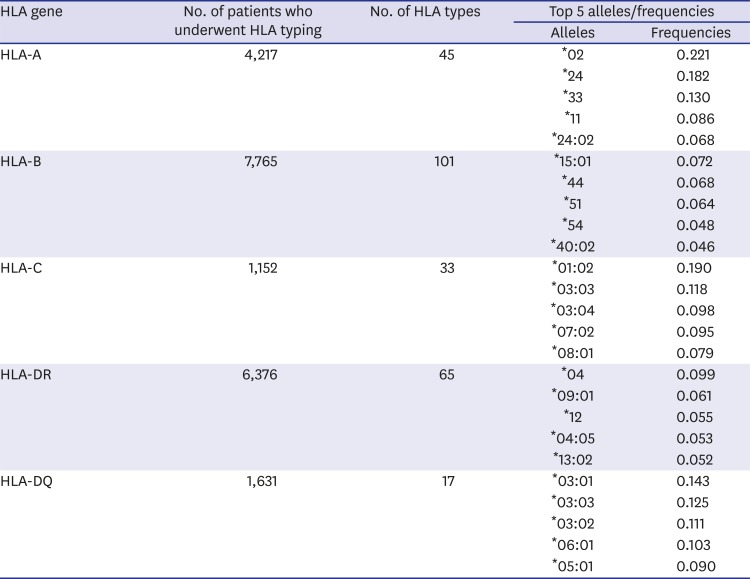
Table 1
Typical Python expressions used to extract the HLA genotype status and clinical variables for each patient
![]()
Table 2
HLA genotype frequencies in the test set extracted by Rule #1 and Rule #2
![]()
Validation of NLP accuracy
Evaluation of the extraction rules was based on the standard evaluation metrics of precision and recall. The performance of the extraction rule was evaluated against a validation set generated by manual curation from the test set by a research nurse. Among the 6,180 reports in the testing set, 1,094 reports were identified to contain a single patient result in the validation set. Among these reports, 1,088 reports (99.5%) for 1,039 patients matched with the set extracted by Rule #1. The precision of Rule #1 (designed to extract reports with HLA typing for a single patient) was 0.997 and the recall was 0.994. There were three patients with data that were not extracted by Rule #1 but that were present in the validation set. All of these cases included names written in English or text with unexpected spaces, and thus were missed by our extraction rules.
The clinical variables including patients' names and indication of HLA typing as well as HLA genotypes, were then extracted by Rule #2, and the results are summarized in Table 3. For the genotype extraction, we developed rules specific for each HLA gene because the resolution of typing and associated descriptions varied for each gene. The average precision of the determined HLA serotype/alleles was 0.976 and the recall was 0.952. The baseline HLA typing frequencies varied among the five HLA genes, with typing for HLA-C being mostly rare and typing for HLA-B being the most common, as observed in rule-development set. As the coverage and variability of rules should be dependent on the number of tests for each HLA serotype/alleles in the rule-development set, HLA-C typing showed the lowest precision (0.892 for serotype and 0.92 for allele) and the lowest recall (0.795 for serotype and 0.821 for allele). By contrast, the top accuracy was obtained for the HLA-DR and DQ genes, which were not the most frequently tested. This is likely related to the fact that these genes were commonly tested using high-resolution techniques, and thus the typing results were represented in a relatively uniform format such as “HLA-gene[Number][Space]*[Number]:[Number]”.
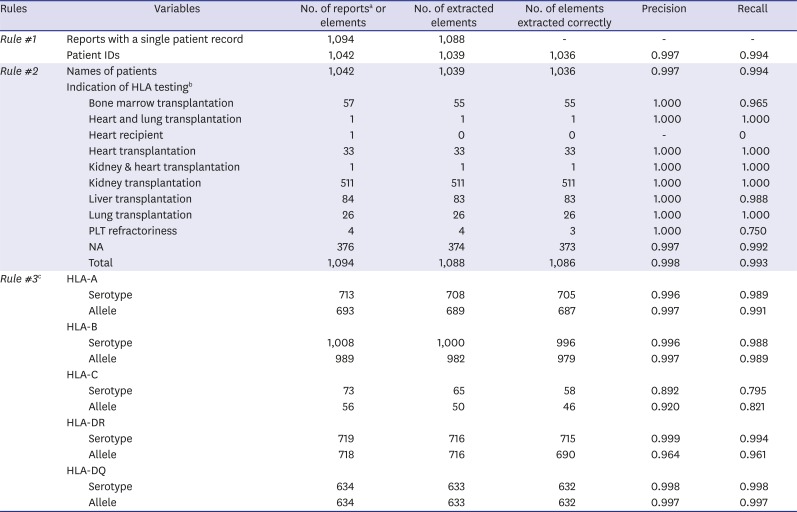
Table 3
Recall and precision for clinical variables and serotype/alleles of HLA genes
![]()
HLA nomenclature mapping
We converted the all of the extracted HLA data into the standard nomenclature format.14 If there was only serotype information available for a particular type of test with low resolution, only the serotype was saved without proceeding to detailed nomenclature conversion. We also developed rules to clean the genotype results by removing escape characters such as “\n” or empty spaces. We also added asterisk (*) or colon (:) to convert the genotypes according to the standard HLA nomenclature format. When there were additional letters or terms in the result, such as “g” or “group” after 4-digit of genotype to indicate a group of identical nucleotide sequences in a peptide-binding domain, we placed these additional symbols in a separate column. This was done for standardization as well as to avoid confusion with the HLA nomenclature in which the “G’” code should follows the first three 3 fields (i.e., six digits) of the allele designation. Instances with other letters that may or may not interfere with the HLA nomenclature, such as “a” or “n”, were subjected to the same process. We used R18 for the development of Rule #3, and the final rules are listed in Table 3.
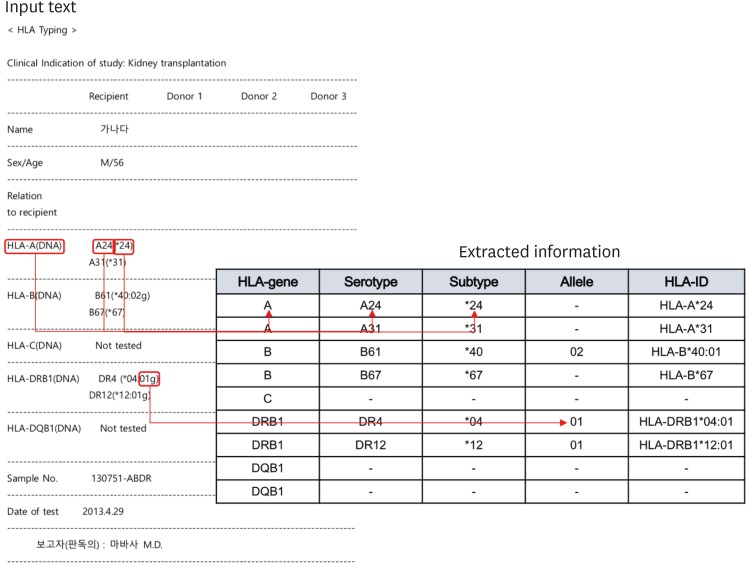
Fig. 3 shows an example of a raw HLA report and the corresponding result for the extraction into the database according to our rules so that the input text-based information and the test results were loaded into the HLA type table under the standardized nomenclature.
Go to :
DISCUSSION
We have provided a useful method to accurately extract HLA data recorded in a free-text format by applying rule-based NLP methods to construct a standardized HLA genotype database. Application of this method to a clinical database showed that serotype (or locus/protein) and allele frequency statistics—which had previously been stored in the form of unrecognizable free-text data—could be confirmed. Convenient access to such background information could help to support valuable clinical decisions for preventing severe adverse drug reactions related to specific HLA alleles. Therefore, we believe that this HLA database will contribute to the utilization of currently underused HLA genotype information to improve the design and development of CDSS and ensure patient safety.
We reviewed all of the HLA genotype mismatches with validation and automatic extraction. There was a patient data that was not extracted by the rules at all for all of the serotype and genotype of five HLA genes and the validation set of him had the correct result. For this case, the row name of the raw data, which represents HLA gene classes, were differently annotated such as “HLA-A antigen” compare to other HLA reports, “HLA-A.” Another case that was not extracted thoroughly was annotated as a donor and written down with unexpected multiple white spaces (indentation). The other cases were all not extracted because they had a complex representation of HLA serotype and genotype with unexpected symbols. For example, a raw report of a false-negative patient was described as the HLA-A serotype as ‘-’ instead of A2.
Based on this application, we demonstrated that rule-based extraction performed reasonably well for processing HLA data in free-text format. The database use in this study was established from various types of HLA tests with a range of resolution levels according to the testing technique and allelic variability in each patient. In current clinical practice, several types of genetic/genomic testing methods are widely adopted, including polymerase chain reaction, fluorescence in situ hybridization, and next-generation sequencing, which all show a wide range of resolution and data representation. One significant aspect of our developed method is that the results of the HLA tests with various level of resolution could all be transformed into a standardized nomenclature, which allowed for obtaining integrated results about the patient without regard to the specific test methods employed in the pilot test. Moreover, the validation results indicated that our approach yielded accurate results when extracting the data for single patients. However, to accurately extract information from the current free-text HLA data, it is essential that an experienced individual directly conducts a review of the results.
The majority of clinical records mostly comprise unstructured data, because these encompass clinical notes made by physicians as well as device-generated reports or imaging/pathology reports, which are largely input in a free-texted or semi-structured formats.19 Unstructured data are continuously produced in clinical settings because the test results are typically generated by different devices in different forms, and physicians typically add their final opinions or remarks in free-text format before loading the reports into the EMR system. This situation is further complicated when physicians use unique, self-made abbreviations. Furthermore, most physicians desire a summary view of the raw data to facilitate making a quick decision in busy clinical practices. Although semi-structured and free-text reports are useful for medical practitioners who are already familiar with such formats, their effective utilization in a CDSS and signal detection can be problematic.20 Therefore, our proposed approach provides a practical solution to convert the underused unstructured genotype data in an EMR to structured and thus reusable data.
The limitations of this study are that we did not perform a utility test of the rule-based named entity recognition system by applying the code to the HLA data generated from other hospitals or organizations. Because the result description pattern of HLA data might vary among institutions, and even between physicians, the performance of the patterns established in this study to extract HLA typing data should be further evaluated in other data sets. We have made this pattern openly available through GitHub and will continuously upbuild the codes. The second limitation is that we only used the regular expression as the NLP method to extract HLA genotype data without considering other trendy methods such as machine learning-based NLP. As shown in Fig. 1, the HLA data in our study did not form a complete sentence or phrase, which was not suitable for traditional text processing. We have adopted regular expressions which have shown that can easily integrate prior knowledge and show reliable performance in clinical text processing, especially in Korean and English mixed EMR data processing.2122 For this reason, we used regular expressions, and the results of the study showed remarkable performance. Lastly, this study process and results is anticipated to proof the long-term reusability, but was not confirmed directly. Because the clinical data generated and stored in EMR is continuously updated and new data are always being added, the extract, transform, and load (ETL) systems should be used to confirm the data reusability and reliability to support the CDSS. However, we constructed the testing set using HLA reports that were recently ordered over one year at SNUH and the performance was found to be reasonable. Therefore, application to newly created data represents the next research challenge. We plan to carry out an advanced modeling study that considers linkages with clinical data and the efficiency of data storage, along with developing data utilization scenarios in the future.
Despite these limitations and remaining challenges, the present study represents the valuable attempt to use data modeling for storing and managing the ever-increasing amount of genomic data contained in EMRs. Although many more developments are expected to complement the process and system, this preliminary applications demonstrates that underused genotype data could be accurately extracted with an NLP method. In particular, for the secondary use of genomic data, it is very important to establish a map according to the resolution of the test and proper nomenclature. Wide adoption of this approach should facilitate access to more reliable data that can be reused for many purposes to the benefit of clinicians in decision-making, and ultimately to the patient.
Go to :
 XML Download
XML Download