

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Gastrointestinal stromal tumors (GISTs) are the most common stromal or mesenchymal subepithelial neoplasms originating in the gastrointestinal tract. Because of their relatively broad morphological distribution, GISTs were formerly called leiomyomas, leiomyosarcomas, and leiomyoblastomas of the gastrointestinal tract, until they were found to have clinical, histopathological, and molecular features that differentiated them from the other soft tissue tumors. GISTs have a characteristic morphology and are generally positive for the CD117 antigen, an epitope of the KIT receptor tyrosine kinase (RTK). GISTs are primarily caused by oncogenic mutations in genes encoding either of 2 RTKs, KIT or platelet-derived growth factor receptor A (PDGFRA) [1]. KIT and PDGFRA are growth factor receptors that are activated by their respective ligands including stem cell factor and PDGF-AA to trigger cellular pathways that upregulate proliferation, downregulate apoptosis, and control cell differentiation, adhesion, and motility under normal conditions. Mutations in KIT and PDGFRA result in the constitutive activation of these cellular pathways, leading to spontaneous proliferation and uncontrolled tumor growth [2]. Different mutations, including point mutations, and deletions and insertions, have been found in the different exons or in different regions of a single exon in KIT (exons 9, 11, 13, and 17) and PDGFRA (exons 12, 14, and 18) genes in GISTs.
Identifying gene mutations in individual tumors is critical to improve the efficacy of cancer therapy by matching targeted drugs to specific mutations. Next-generation sequencing (NGS) technologies have revolutionized cancer genomics research by providing an unbiased and comprehensive method for detecting somatic cancer genome alterations. Targeted NGS with a gene panel is a powerful and practical approach as it enables the analysis of high-yield genes or genomic regions with relatively rapid turnaround time, low DNA input, and low cost [345].
Several targeted NGS panels are commercially available, although most of them are designed to cover the important alterations in various cancers. The Ion Torrent™ Oncomine Focus Assay (OFA) is a targeted NGS assay that enables the analysis of over 1,000 biomarkers across 52 key solid tumor genes that are well characterized in published literature and associated with current oncology drugs [67]. This assay comprises 2 separate panels (DNA and RNA) that are designed to interrogate hotspot mutations (35 genes), copy number variations (19 genes), and gene fusions (23 genes). Together, these 2 panels can identify current actionable genetic variants and potential future targets for personalized therapy.
In this study, targeted NGS assay with an OFA panel was performed for the genetic characterization of molecular targets in 30 Korean patients with GIST. In addition, we assessed whether our molecular analysis could be considered as surrogate markers when compared to risk assessment criteria.
MATERIALS AND METHODS
Specimens
The study protocol was approved by the Institutional Review Board of The Catholic University of Korea, including written informed consent for clinical and molecular analyses (DC18SESI0113). A total of 30 formalin-fixed and paraffin-embedded (FFPE) specimens of GIST were included in the study. The specimens were obtained by surgical resection between April 2014 and August 2018 at the Daejeon St. Mary's Hospital, Republic of Korea. Specimens were selected based on the archival histopathological report and subsequent review by experienced pathologists. Tumor content was in the range of 50%–90%.
Risk assessment
Because the tumor site was located only in the stomach in all 30 patients with GIST, the National Institutes of Health (NIH) GIST Consensus Criteria was applied for GIST risk assessment. The criteria utilize 2 clinical pathological factors, tumor size and mitotic count, and stratify recurrence risk as very low, low, intermediate, or high. Several reports of patients with localized GIST treated with surgery alone have confirmed the prognostic value of both tumor size and mitotic count [89].
DNA isolation and quantification
Genomic DNA was isolated from the FFPE samples using the RecoverAll Total Nucleic Acid Isolation Kit (ThermoFisher Scientific, Waltham, MA, USA) per the manufacturer's instructions after de-paraffinization and extraction of 1–2 mm thick paraffin sections in xylene. Amplifiable genomic DNA was quantitatively assessed using a Qubit 2.0 Fluorometer (ThermoFisher Scientific), a Qubit dsDNA High Sensitivity (HS) Assay Kit (ThermoFisher Scientific), and TaqMan RNase P Detection Reagent Kit (ThermoFisher Scientific) as appropriate.
Library preparation
DNA libraries were constructed using the Ion AmpliSeq Library Kit 2.0 (ThermoFisher Scientific) according to the manufacturer's recommendations. The Oncomine Focus DNA Assay (ThermoFisher Scientific) was used to generate sequencing libraries using a total of 10 ng input genomic DNA per sample. This DNA panel is specifically optimized for detection of hotspots, single nucleotide variants (SNVs), insertion and deletions (Indels), and copy number variants (CNVs) across the following genes commonly implicated in human cancers and relevant to the targeted treatment of solid tumors: AKT1, ALK, AR, BRAF, CDK4, CTNNB1, DDR2, EGFR, ERBB2, ERBB3, ERBB4, ESR1, FGFR2, FGFR3, GNA11, GNAQ, HRAS, IDH1, IDH2, JAK1, JAK2, JAK3, KIT, KRAS, MAP2K1, MAP2K2, MET, MTOR, NRAS, PDGFRA, PIK3CA, RAF1, RET, ROS1, and SMO for hotspot mutations; ALK, AR, BRAF, CCND1, CDK4, CDK6, EGFR, ERBB2, FGFR1, FGFR2, FGFR3, FGFR4, KIT, KRAS, MET, MYC, MYCN, PDGFRA, and PIK3CA for focal CNV gains. The Oncomine Focus RNA Assay is also available in the Oncomine Focus Panel, however, gene fusions were not analyzed in the assay because of several actionable gene fusions such as FGFR1-HOOK3, FGFR1-TACC1, and ETV6-NTRK3 fusions for GIST were not included. Unique Ion Xpress Barcode 1–16 and Ion P1 Adapters (ThermoFisher Scientific) were ligated to the amplicons and subsequently purified to ensure that each individual sample had a unique ID. The final amplicon libraries were then amplified, purified, and equalized up to 100 pM using AMPure XP Reagent (Beckman Coulter, Indianapolis, IN, USA).
Semiconductor sequencing
Six uniquely barcoded library samples were pooled per run for sequencing on an Ion 318 v2 chip (ThermoFisher Scientific). The Ion Chef System (ThermoFisher Scientific) was used with the Ion PGM Hi-Q Chef Kit (ThermoFisher Scientific) for fully automated template preparation and Ion 318 v2 chip loading. Single-end sequence analysis was performed using the Ion PGM Hi-Q Sequencing Kit on the Ion Torrent Personal Genome Machine (Ion PGM) (ThermoFisher Scientific) for 200 base-read sequencing.
Variant calling and data analysis
Raw data from the DNA panel were collected and processed to generate sequence reads and trimmed using the Ion Torrent platform-specific pipeline software. Removal of polyclonal and poor signal-profile reads as well as 3′ quality trimming of the reads was performed using Torrent Suite Assay Development Mode v5.0 (ThermoFisher Scientific). The reads were aligned to the reference genome (human genome hg19) and Ion Reporter v5.1 software package (ThermoFisher Scientific) was used for data analysis of DNA panel. ThermoFisher recommends 500x coverage to detect somatic mutations using their AmpliSeq technology; a 500x coverage cut off was applied to all analyses in this study. As a result, the target regions with >500× demonstrated sufficient and uniform amplification and sequencing coverage, with mutant alleles detected at 5% allele frequency. Briefly, the ‘Oncomine™ Focus - 520 - w2.4 - DNA - Single Sample’ automatic workflow in Ion Reporter was used to identify and annotate the SNVs, Indels, and CNVs from the OFA. This workflow has preconfigured Torrent Variant Caller (TVC) parameter settings: Min Allele Freq (frequency cutoff for supporting a variant), SNV 0.04, InDel 0.07, Hotspot 0.03; Min Coverage (total coverage required for reads or no-call), SNV 15, InDel 15, Hotspot 15; Strand Bias (proportion of variant alleles comes overwhelmingly from one strand) SNV 0.96, InDel 0.9, Hotspot 0.96 for SNVs and Indels calling, and 5% CI CNV ploidy ≥gain of 2 over normal was used for CNV calling.
Bioinformatic analysis and experimental validation
To distinguish between somatic and germline mutations, the detected mutations were compared to the variants in the Exome Aggregation Consortium (http://exac.broadinstitute.org/), 6,500 exomes of the National Heart, Lung and Blood Institute (NHLBI) GO Exome Sequencing Project (http://evs.gs.washington.edu/EVS/), and 1,000 Genomes Project [10]. To estimate recurrent mutations in GIST, ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and the COSMIC (version 88) (https://cancer.sanger.ac.uk/cosmic) databases were used. Additionally, direct sequencing was performed to confirm some of the detected mutations (mutant allele burden >20%) other than hotspot mutations with no COSMIC ID.
Statistical analysis
Fisher's exact test was used to compare the mutation profiles and histopathological findings by NIH risk assessment. Multivariable logistic regression was used to calculate adjusted odds ratio for NIH risk assessment for these variables including mutation profiles. Survival and disease-free survival analyses were not performed because all patients were alive and disease-free during the study period. All statistical analyses were performed using MedCalc Statistical Software Version 17.6 (MedCalc software, Ostend, Belgium). Statistical significance was set at P<0.05.
RESULTS
The cohort comprised of 50% (15/30) male and 50% (15/30) female Korean patients with a median age at 62 years (range, 41–79 years) with GIST. During the study period (median follow-up period 865 days) there was no recurrence or death from any cause. Four out of the 30 patients received adjuvant imatinib treatment. Immunohistochemistry (IHC) results for c-kit or discovered on GIST-1 (DOG1) were available for 28 out of the 30 GISTs: All 28 were positive for c-kit (28/28) and DOG1 (22/22), whereas there was no negative result for IHC of c-kit or DOG1. The clinicopathologic features of the 30 GIST patients are summarized in Supplementary Table 1. After applying stringent parameters for reliable variant calling (allele burden >4%, coverage depth >500×) and after filtering out potential raw base calling errors, 43 hotspot/other likely pathogenic variants (26 hot spots and 17 other variants; 33 SNVs, 8 Indels, and 2 amplifications) in 16 genes were identified in 26 out of the 30 GISTs. Other than hotspot mutations with >allele burden of 20% were confirmed by direct sequencing. Only one patient out of the remaining 4 GISTs without any alterations received the adjuvant imatinib treatment. KIT variants were most frequent (44%, 19/43), followed by 6 variants in PIK3CA, 3 in PDGFRA, 2 each in JAK1 and EGFR, and one 1 in AKT1, ALK, CCND1, CTNNB1, FGFR3, FGFR4, GNA11, GNAQ, JAK3, MET, and SMO. Based on the mutation types, most of the variants were missense mutations (60%, 26/43), followed by 8 frameshifts, 6 nonsense, and 1 stop-loss, and 2 amplifications (Fig. 1). Previous studies showed that functional SNVs may contribute to the development of GISTs [121112]. Fig. 2 illustrates the incidence of SNVs in combination with other mutations in this study. The results showed that 81% (21/26) of samples had at least 1 or more SNVs, 38% (10/26) had at least 2 or more SNVs, while 69% (18/26) of samples incurred no Indels. A detailed list of frameshift, stop-loss, nonsense, and missense mutations, and amplifications identified in this study is provided in Tables 1 and 2.
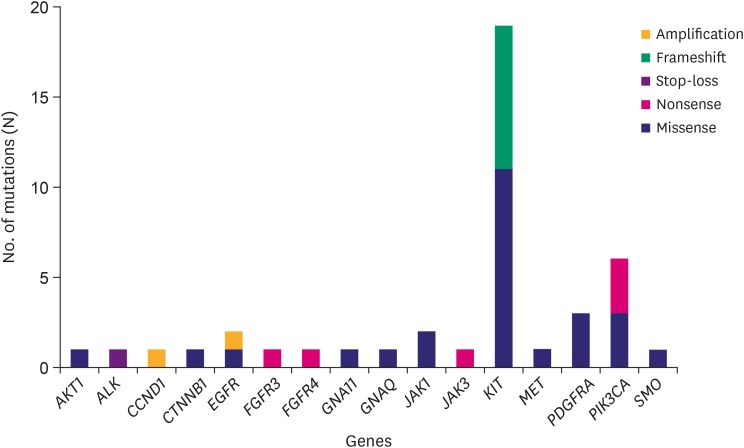
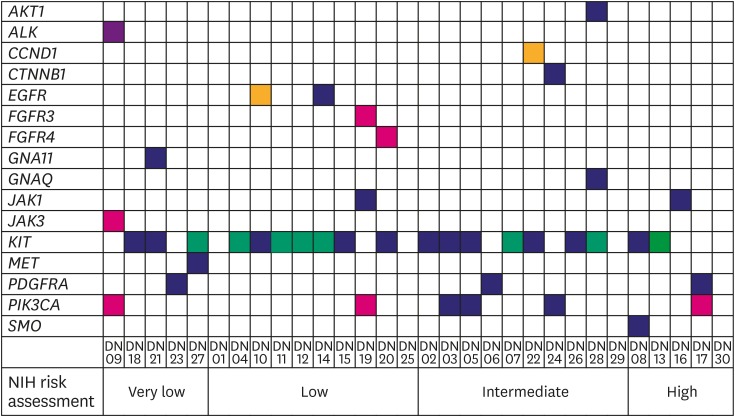
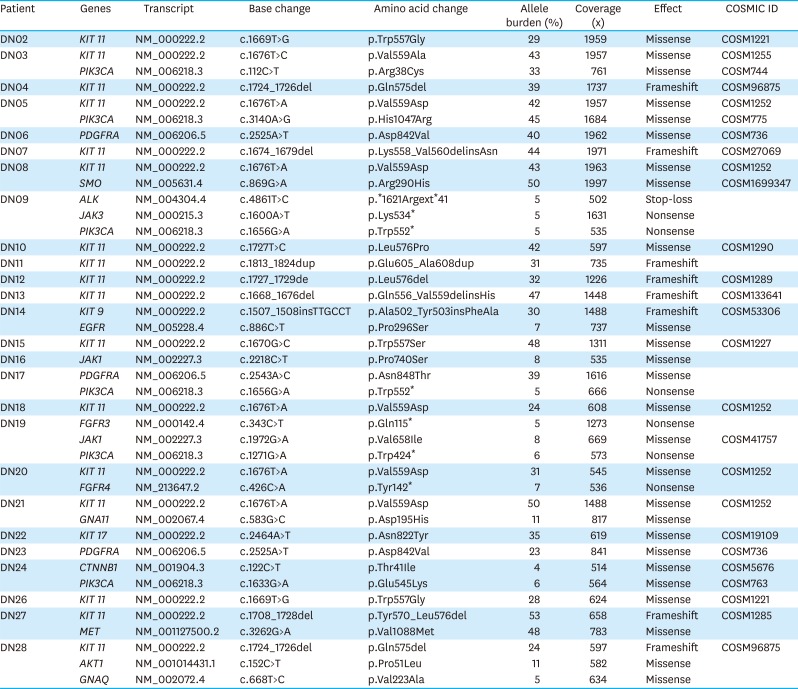
Fig. 1
Frequencies of somatic mutation types detected in the various genes by the Oncomine Focus DNA Assay in 26 GISTs. Gene identities are depicted on the x-axis, and frequency of mutations on the y-axis.
GIST = gastrointestinal stromal tumor.
![]()
Fig. 2
Distribution of somatic mutation types detected for each patient by Oncomine Focus DNA Assay in 26 GISTs. Individual patients are depicted on the x-axis, and identity of genes on the y-axis. Yellow box, amplification called; green box, frameshift mutation called; violet box, stop-loss mutation called; red box, nonsense mutation called; indigo box, missense mutation called; white box, no mutation called.
GIST = gastrointestinal stromal tumor; PDGFRA = platelet-derived growth factor receptor A; NIH = National Institutes of Health.
![]()
Table 1
Results of somatic mutations identified by Oncomine Focus DNA Assay in 26 gastrointestinal stromal tumors
![]()

KIT and PDGFRA mutations in GISTs
Accumulating evidence indicates that activating mutations of KIT or PDGFRA are the initiating event in GISTs. Activation of KIT or PDGFR leads to downstream signaling in the PI3K, Ras, and JAK/STAT pathways, resulting in increased cell proliferation and inhibition of apoptosis [1314]. Accordingly, 19 (63%) of the 30 GISTs sequenced in this study had KIT gene mutations; mutations in exons 9 (n=1), 11 (n=17), and 17 (n=1) were found located in regions corresponding to the transmembrane and cytoplasmic domain but not along the extracellular domain. Mutations in exon 11 (89%, 17/19), which encodes the regulatory domain of the enzyme, were found to carry missense mutations in 10 patients (59%, 10/17) and frameshift mutations in 7 patients (41%, 7/17). Exon 9, which encodes the immunoglobulin-like C2-type 5 domain, was mutated only in one patient (5%, 1/19) with an insertion of c.1507_1508insTTGCCT resulting in p.Ala502_Tyr503insPheAla. Exon 17, which encodes the kinase activation loop, was also mutated only in one patient (5%, 1/19) with a substitution of c.2464A>T resulting in p.Asn822Tyr. These data support the critical role of KIT mutations in the development and progression of GISTs.
Our results also showed that 3 (10%) of the 30 GISTs sequenced in this study had PDGFRA gene mutations: exon 18 (n = 3) was found to be mutated in the cytoplasmic domain. Mutations in exon 18 (100%, 3/3), which encodes the kinase domain of the enzyme, included missense mutations (2 p.Asp842Val and 1 p.Asn848Thr) in the 3 patients (59%, 10/17).
Influence of mutation profiles on NIH risk assessment
To estimate the correlation of NIH risk assessment with selected histopathological findings and mutation profiles, the 4 criteria in the NIH risk assessment were categorized into 2 distinct groups: ‘very low & low’ and ‘intermediate & high’ (Fig. 2). Based on Fisher's exact test, there was no statistical significance between NIH risk assessment and selected histopathological findings or mutation profiles except for necrosis. Patients in the ‘intermediate & high’ (53%, 8/15) group had an increased number of necrotic cells, compared to the patients in the ‘very low & low’ (0%, 0/15; odds ratio, 2.143; 95% confidence interval, 1.247–3.681; P=0.002) group. However, multivariable logistic regression analysis demonstrated that the ‘intermediate & high’ criteria for NIH risk assessment were not associated with necrosis or the histopathological findings and mutation profiles (Table 3).
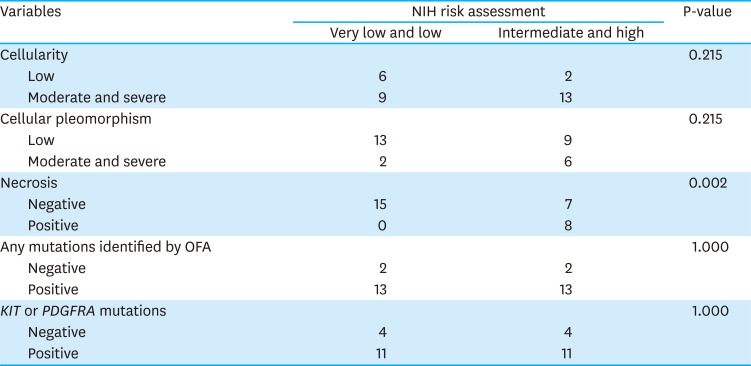
Table 3
Correlation of NIH risk assessment with select histopathological features and mutation profiles
NIH = National Institutes of Health; OFA = Oncomine Focus Assay; PDFGA = platelet-derived growth factor receptor A.
![]()
DISCUSSION
GISTs are mesenchymal neoplasms that arise primarily from within the muscular wall of the stomach and small intestines; they rarely occur in extra-intestinal locations. The aggressiveness of the tumor is correlated with tumor size, mitotic activity, and anatomical origin [15]. GISTs frequently harbor activating gene mutations in either KIT or PDGFRA genes and thus are highly responsive to several selective tyrosine kinase inhibitors [1216]. Surgical resection is currently the only curative treatment for localized GIST and constitutes over half of potentially curative GIST treatments. However, the clinical course of GIST ranges from that of a tumor cured by surgical resection to that of a locally advanced or even widely metastatic, and ultimately fatal, disease. This clinicopathologic heterogeneity is paralleled by an underlying molecular diversity: the majority of GISTs are associated with spontaneous activating mutations in KIT, PDGFRA, or BRAF genes, while additional subsets are driven by genetic lesions (often inherited) in NF1 or components of the succinate dehydrogenase enzymatic complex [1718]. Routine genotyping has become an integral part of the management of GISTs undergoing tyrosine kinase inhibitor therapy [15].
In this study, we aimed to identify and evaluate genetic mutations in GIST for selective molecular targets using targeted NGS with the OFA panel. This approach identified 19 KIT and 3 PDGFRA mutations as missense or frameshift mutations in 22 different patients with GIST. Most KIT mutations (89%, 17/19) were in exon 11 of the KIT gene; only one mutation each was detected in exon 9 and 17 (Fig. 3). Gain-of-function mutations in KIT result in a growth advantage by constitutive, ligand-independent activation of the RTK [19]. While most GISTs are heterozygous for a given mutation, in around 15% of the tumors, the remaining wild-type KIT allele is lost, and this is associated with malignant behavior, increased mitotic activity, and topoisomerase II expression [20]. The most frequent hotspot mutation in KIT is detected in exon 11; other mutations are less frequently detected in exon 9 and rarely in exon 13 and exon 17 [21]. Importantly, the presence of KIT del-ins557/558 is an important prognostic factor for poor outcome in comparison with other KIT exon 11 mutations, KIT exon 9 and PDGFRA exon 18 mutations, even in tumors classified as non-high-risk ([very]low and intermediate) that originate from the stomach [22]. In this study, 2 KIT del-ins557/558 and 2 missense mutation at the position 557 were identified. Prognostic evaluations were difficult because the patients were alive during the study period.
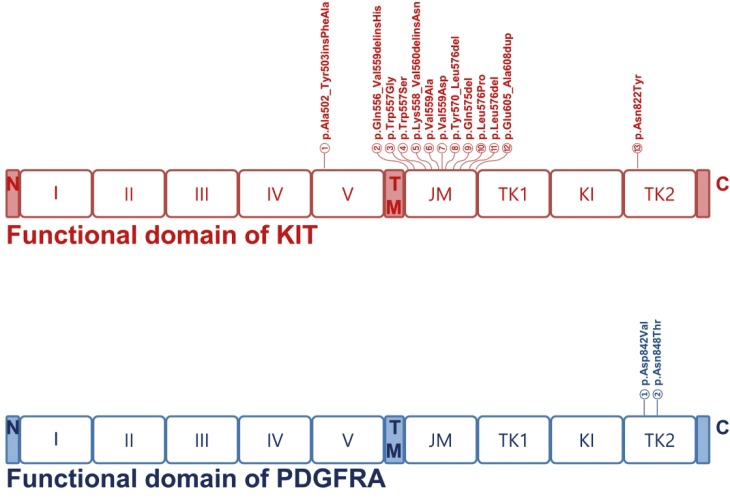
Fig. 3
Distribution of mutations in KIT and PDGFRA functional domains. Somatic mutations in KIT and PDGFRA identified in our study are shown. Boxes represent functional domains: I–V, 5 immunoglobulin-like domain; TM, transmembrane domain; JM, juxtamembrane domain; TK1, tyrosine kinase domain 1; KI, kinase insert domain; TK2, tyrosine kinase domain 2. The type of mutation detected within the domains are indicated above.
GIST = gastrointestinal stromal tumor; PDGFRA = platelet-derived growth factor receptor A.
![]()
PDGFRA and KIT mutations are mutually exclusive and activate similar downstream signal transduction pathways. However, PDGFRA-mutant GISTs are almost exclusively of gastric origin (90%–93%), which is prognostically favorable [12]. The most prevalent genotype is the p.D842V substitution involving the second kinase domain (which corresponds to exon 17 of KIT), which is detected in 60%–65% of all PDGFRA mutated tumors [23]. In this study, 2 patients (DN06 and DN23) carried an exon 18 D842V substitution of the PDGFRA only and were classified as very low and intermediate by NIH risk assessment, respectively. A previous study suggested that GISTs with PDGFRA exon 14 mutations represent a subset of clinically favorable gastric tumors (exclusively gastric location) with almost exclusively epithelioid morphology [24]. However, there was no exon 14 alteration of the PDGFRA in this study.
On the other hand, GISTs without activating mutations in KIT and/or PDGFRA genes tend to have 2 or more mutations (75%, 3/4), compared to GIST with KIT and/or PDGFRA mutations (45%, 10//22). Among them, co-occurrence with PIK3CA mutations was dominant: Detection of PIK3CA mutations in large or metastatic KIT-mutant GISTs may suggest that PIK3CA-mutant clones have a proliferative advantage during disease progression. Tyrosine kinase inhibitors have been successfully used in GIST treatment. However, resistance frequently develops due to secondary KIT mutations or activation of downstream signaling pathways, such as the PI3K/AKT/mTOR pathway. Genotyping of PIK3CA in GISTs may help to differentiate between primary and metastatic tumors with the potential to develop resistance to tyrosine kinase inhibitors and guide therapies with PI3K inhibitors [25].
Interestingly, 2 amplifications, one each in the CCND1 and EGFR genes were identified in GISTs with KIT missense mutation. In previous studies, amplifications were reported for CMYC in 3 of 90 (3.3%), for MDM2 in 5 of 94 (5.3%), for EGFR1 in 5 of 94 (5.3%), and for CCND1 in 7 of 79 (8.9%) evaluable cases. Among them, MDM2 and CCND1 amplifications were associated with clinical and histological malignancy [26]. On the other hand, there was no correlation between EGFR gene amplification or EGFR protein overexpression with GIST [27]. In our study, the EGFR amplification was detected in a low risk, whereas the CCND1 amplification was detected in an intermediate by NIH risk assessment case.
Identifying single or combinations of mutations with the aim of delivering individualized treatment with a single or combination of target agents has been an effective strategy for cancer therapy [2]. Approximately 85%–90% of GISTs harboring KIT or PDGFRA mutations benefit from imatinib treatment before or after surgery and in the setting of unresectable/metastatic disease [28], except for tumors with some specific mutations such as PDGFRA exon 18 D842V [29]. The remaining 10%–15% of GISTs without KIT or PDGFRA mutations are classified as wild-type GIST. These tumors do not respond to imatinib. In this group, several mutations have been identified in genes including those encoding succinate dehydrogenase (SDH) complex subunits, neurofibromatosis type 1, BRAF, and other genes [30]. European Organisation for Research and Treatment of Cancer (EORTC) and Scandinavian Sarcoma Group (SSG) trials further suggested that adjuvant imatinib treatment should be carefully applied to high-risk patients and that the tumor genotype should also be taken into consideration. For example, in the advanced/metastatic setting, the PDGFRA exon 18 D842V mutated GISTs do not benefit from imatinib [29] and a higher dose of imatinib (800 mg daily) is recommended by some institutions for KIT exon 9-mutated GISTs [31]. Evaluation of specific mutations can provide information for a specific tailored therapy. Further trials led to the approval of 2 more drugs by the FDA, sunitinib and regorafenib, which expands options for GIST treatment following failure of imatinib.
Unfortunately, there are 2 major limitations in our study. Due to the inherent underrepresentation of genes in the OFA, critical genes could not be investigated. The OFA does not cover the presence of significantly mutated driver genes that were previously identified (TP53, ARID1A, and CDH1) and some new ones (MUC6, CTNNA2, GLI3, RNF43, and others) associated with gastrointestinal tumor/carcinoma [32]. In fact, the OFA used in this study was too small gene panel for excavation of the complicated genetic alterations in GIST. Thus, a combination of the oncomine comprehensive panel and the Ion S5XL platform (ThermoFisher Scientific) would be better suited for application in routine clinical NGS test for solid tumors [33]. Second, it is possible that no calls of mutations in 4 GISTs with wild-type were due to low tumor purity. Both regular prospective and retrospective quality management processes and adequate designation to enrich tumor cellularity by pathologists in the molecular diagnostics laboratory can reduce the risk for a false negative result. The NGS platform with higher analytic sensitivity can detect mutations in specimens with lower tumor cellularity [34]. Finally, in our study, the relapse-free or disease-free survival analysis was not applicable due to the small sample size (n=30) and short median follow-up duration.
In conclusion, our study confirms the utility of the Ion Torrent sequencing platform with an OFA panel to efficiently identify KIT and PDGFRA mutations associated with GISTs, and other gene mutations associated with solid tumors. These findings may provide a genetic basis for developing new GIST therapeutic agents specifically targeting these gene mutations. As more experience and information are gained from the NGS, it is necessary to expand our understanding of the sensitivity of individualized therapies to specific mutations.
 XML Download
XML Download