

 PDF
PDF ePub
ePub Citation
Citation Print
Print



Familial Alzheimer disease (AD) is caused by mutations in the PSEN1, PSEN2 and APP genes. In rare cases, mutations in ABCA7 and SORL1 also show familial AD, depending on their positions in the proteins.12 About 5% of early onset AD (EOAD) are caused by mutations of these 5 genes.3 The clues about having causal gene mutations in EOAD are the age of onset and number of EOAD patients in their families. If EOAD patients have one or more relatives with EOAD, 77% of their families have the causal gene mutations.4 In sporadic cases, the causal gene can be identified at a very low rate of 17.9% for patients of age at onset under 50 and 1.2% for those over 50.4 Familial EOAD cases have been reported due to a loss of function mutations that seriously alter the function of the ABCA7 gene.2 The mutations in genes other than ABCA7 make it difficult to determine pathogenicity. In this letter, we report a case of the R278I mutation in PSEN1, which has not been reported in Korean familial EOAD patients.
A 49-year-old man visited our dementia center complaining of memory impairments. According to the patient's report, he felt mild forgetfulness for about 10 years. In 2018, he began to easily forget the stories that he heard. Sometimes, he could not remember what he had done at work. When he talked with friends, he often could not remember the important previous events. His friends advised him to visit clinics. As a computer programmer, there is no problem in working. Driving, money management, hobbies and social relations are maintained as before. No neurological findings were observed. According to neuropsychological tests, his clinical diagnosis was multiple domain amnestic mild cognitive impairment (MCI). He remembered that his father had dementia in his early sixties, and his brother also developed dementia in his early fifties. His mother has still normal cognition to date. The patient has 2 sisters who are 58 and 55 years old, and neither of them have ever complained of memory impairment (Fig. 1). No atrophy was observed on magnetic resonance imaging (MRI) and there was no subcortical ischemic change. On the Vizamyl TM (flutemetamol F18; GE healthcare, Chicago, IL, USA) amyloid positron emission tomography, there was amyloid deposition in the cerebral cortex. The Institutional Review Board approved the genetic study of the family (IRB approval number: 2016-02-010). We performed whole exome sequencing and found a mutation in the proband, which changed guanine into thymine of hg19 73664802th nucleotide in chromosome 14. The mutation changed arginine (R) into isoleucine (I) in 278th amino acid of PSEN1. The R278I mutation is found also in his demented brother and sister, who has normal cognition to date (Fig. 2).
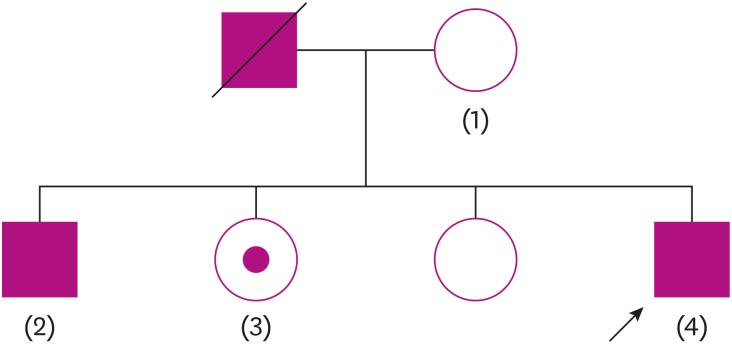 | Fig 1The pedigree of the family in the case. The arrow indicates proband, and the subjects with numbers were sequenced.
|
 | Fig. 2Integrative genome browser (https://software.broadinstitute.org/software/igv/) view. The proband, sister and brother had chr14:73664802:G:T (p.Arg278Ile; hg19) mutation. The mother, who had normal cognition, did not have the mutation.
|
This familial case is the first reported R278I mutation of PSEN1 in Korea. However, the mutation was reported in one family in the foreign country.5 The reported patients showed cognitive impairment accompanied by language impairment. The clinical diagnosis was progressive non-fluent aphasia. A similar R278T mutation of PSEN1 was reported to be associated with spastic paraparesis.6 In contrast, the patient in this letter has not shown any neurological symptoms. There may be three possibilities. Now, proband is in the MCI stage and different symptoms may develop later. There may also be unknown genetic cofactors related with diverse phenotypes. Finally, epigenetic and environmental factors can cause various symptoms.
The 278th codon is located in the sole cytoplasmic active domain for pathogenic mutations. Most of pathogenic mutations of PSEN1 are located in the transmembrane domains.7 The 278th amino acid of PSEN1 lies in the cytoplasmic domain, which forms the gamma-secretase complex.8 The mutations in that domain are expected to alter the function of gamma-secretase and generate Aβ42 amyloid.
The white matter hyperintensities (WMH) are related with locations of PSEN1 mutations. Although the post-200th codon mutations are related with occipital WMH,9 the proband's MRI did not show the WMH. The severity of WMH are correlated with age. Therefore, we cannot determine if the R278I mutation of PSEN1 tend to cause WMH, yet.
The missense mutations are the most common mutations in humans.10 The clinical information from family studies are strong evidence to determine pathogenicity of the mutations.11 The foreign free AD mutation database (https://www.molgen.ua.ac.be/ADMutations/) was closed and we need to pay in order to use the database. In Korea, there must be only a limited number of families with PSEN1 mutations. Therefore, discovering mutations for Korean EOAD and reporting the results will help in diagnosing EOAD patients.
 XML Download
XML Download