

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
The mammalian heart is highly oxidative, consuming high levels of oxygen to generate adenosine triphosphate (ATP) for myocyte contraction and relaxation.1) Mitochondria are highly dynamic cell organelles that take part in a wide-range of functions. In cardiac myocytes, mitochondria are crucial for heart function through oxidative ATP generation. In addition to energy production, mitochondria play a role in fatty acid synthesis, amino acid production, heme synthesis, iron-sulfur cluster biogenesis, and act as a signaling hub for innate immunity and cell death.2) Mitochondrial dysfunction leads to energetic dysfunction, oxidative stress, calcium dysregulation, and cardiomyocyte death.3) Therefore, mitochondrial dysfunction is currently being considered as a potential therapeutic target.
To achieve mitochondrial homeostasis and maintain a healthy mitochondrial status, cardiac myocytes have quality control mechanisms known as mitochondrial quality control (MQC) systems.4) For the maintenance of mitochondrial protein homeostasis, mitochondria contain diverse chaperons and proteases that facilitate communication with other organelles, termed mitochondria-cytosol-nucleus crosstalk.5) If damages in the mitochondria are not rescued, the disrupted mitochondria are engulfed by an autophagosome for lysosome degradation.6)
Recent studies revealed the significant role of mitochondrial dysfunction in heart disease and the emergence of mitochondrial function restoration as a novel therapeutic target.7)8)9) In this review, we discuss the MQC systems and their role in cardiac protection and heart failure development. We then describe novel drug candidates (urolithin A and spermidine) for the treatment of cardiovascular disease (CVD) by maintaining mitochondrial homeostasis and restoring mitochondrial function.
MITOCHONDRIAL QUALITY CONTROL SYSTEMS
Mitochondrial proteostasis
The mitochondria are a central hub of cellular metabolism and signaling. Therefore, keeping the proteomic integrity in the mitochondria is crucial for cell survival.10) To protect the heart under stressful conditions, protein homeostasis is maintained through several mechanisms or “proteostasis” within the mitochondria. Mitochondrial proteostasis includes mitochondrial-localized chaperons and proteases, degradation of bulk mitochondrial organelles, mitochondrial-nuclear communications, and transcellular signaling (e.g., mitokine signals) (Figure 1).11)12)13) To protect the mitochondrial proteome against reactive oxygen species (ROS) damage and misfolded protein toxicity, mitochondria have localized protective machines such as chaperones, antioxidant enzymes, and proteases within mitochondria. The key chaperones include mitochondrial Hsp70 (mtHsp70), mtHSP90, and the large chaperonin Hsp60/10 complex, which fold nascent polypeptides or repair misfolded proteins.14)15)16) Superoxide dismutase enzymes such as SOD2 in the mitochondrial matrix and SOD1 in the intermembrane space (IMS) protect the mitochondria from ROS damage.17) Several proteases in the mitochondrial matrix (e.g., LonP1 and ClpP) and within the IMS (e.g., m-AAA and i-AAA) degrade damaged mitochondrial proteins.18) In addition, mitochondrial dynamic processes such as mitochondrial fusion and fission also play a role in maintaining healthy mitochondria. Mitochondrial fusion dilutes the effects of small amounts of damage19) while mitochondrial fission separates damaged mitochondria from healthy mitochondria. Segregated mitochondria are subsequently degraded by mitophagy.20)
Figure 1
Mitochondrial quality control system. Under normal condition, ATFS-1 and PINK1 proteins goes into mitochondria and are degraded. After mitochondrial stress, disrupted mitochondrial integrity and function induces mitochondrial stress responses for restoring mitochondrial and cellular homeostasis. ATFS-1 traffics to the nucleus and activates transcriptional responses to recover mitochondrial function. Damaged mitochondria are marked by PINK1 and removed by mitophagy pathway.
ATFS = activating transcription factor associated with stress; FGF = fibroblast growth factor; GDF = growth differentiation factor; PINK1 = PTEN-induced kinase 1; Ub = ubiquitin; ROS = reactive oxygen species.
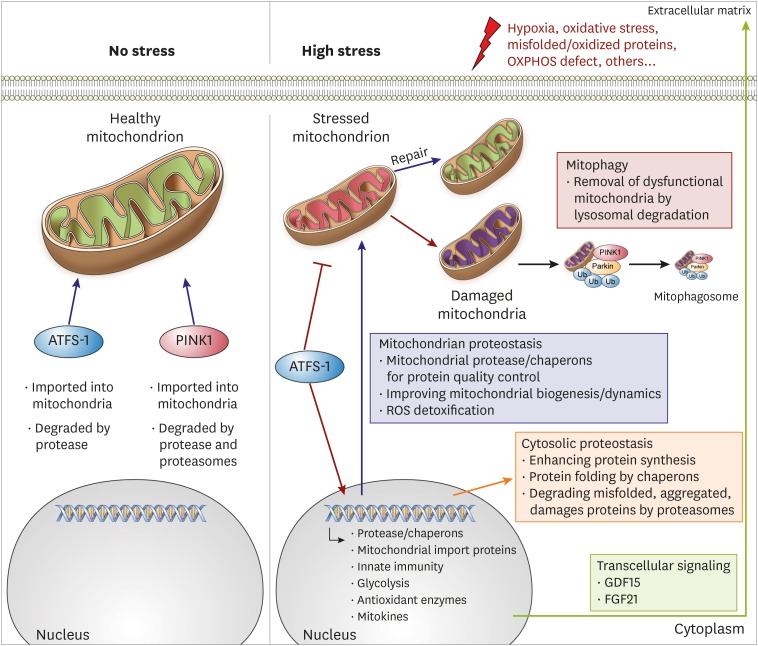
Only 13 proteins are encoded within the mtDNA, while 1,000–1,500 resident mitochondrial proteins are encoded by nuclear DNA.21) This implies that most mitochondrial proteins should be correctly folded within the cytoplasm and imported into the mitochondria.22) Therefore, vast communication between the mitochondria and other cellular compartments is expected. The mitochondrial unfolded protein response (UPRmt) and mitophagy are two major mechanisms for maintaining mitochondrial proteostasis, which include intracellular communication, in the context of the cell.22)
Unfolded mitochondrial protein response
Poor quality mitochondria increase cellular oxidative stress and degenerative apoptosis signals leading to cell death.23) Disrupted mitochondrial proteostasis and dysfunction initiates a retrograde signaling pathway from the mitochondria to the nucleus. This retrograde signaling, known as UPRmt, initiates a transcriptional program to relieve mitochondrial stress.24) The UPRmt is a coordinated response mediated by mitochondrial-nuclear communications, triggered by mtDNA depletion or protein misfolding in the mitochondrial matrix.25) Although this pathway was discovered in mammalian cells, parts of its regulation mechanism have been discovered in worms. In Caenorhabditis elegans, mitochondrial proteotoxicity reduces the mitochondrial import efficiency of a stress activated transcription factor, activating transcription factor associated with stress (ATFS)-1, which is the potential transcription factor mammalian ortholog of activating transcription factor (ATF) 4, ATF5, and C/EBP homologous protein.22) Under physiologic condition, ATFS-1 goes into the mitochondria and is degraded by a Lon protease.25) As a result of mitochondrial perturbation, mitochondrial import is compromised and allows ATFS-1 to be trafficked to the nucleus. Once there, it induces hundreds of genes associated with proteases, antioxidant enzymes, and genes involved in mitochondrial dynamics, protein import, and cellular metabolism.22)
Interestingly, there are significant differences in the mammalian UPRmt signaling cascade compared to C. elegans. For example, C. elegans mitochondrial ClpP protease has a central role generating peptides and acting as a retrograde messenger by proteolyzing misfolded proteins. However, genetical ClpP depletion in the hearts of mice did not affect UPRmt signaling.26)
In the mammalian mitochondria, ATF5 is a proposed mediator of UPRmt.27) In previous studies, pharmacological induction of UPRmt was found to play a role in protecting the heart of wild-type (WT) mice after ischemia-reperfusion (I/R) injury but was not involved in protecting ATF5 knockout mice hearts. Further RNA sequencing results revealed that UPRmt induced ATF5-dependent pathway contributes to cardioprotection.28) Another possible additional main regulator of UPRmt in mammals is ATF4. By treating HeLa cells with 4 therapeutic drugs (doxycycline, actinonin, FCCP, MitoBloCK-6) altering mitochondrial proteostasis and performing multi-omics analyses, Pedro M. et al. reported that ATF4 coordinates mitochondrial stress response.29) ATF4 is essential to maintain cell proliferation and protect the cell against mitochondrial stress; however, it is not involved in mtDNA metabolism.29)
Mitophagy
Mitophagy is a critical MQC mechanism in cardiac myocytes. Impaired mitophagy leads to accumulation of aberrant mitochondria, loss of myocytes, and contractile dysfunction.30) Mitophagy is defined as mitochondrial degradation through the macroautophagy pathway.23) This removal process is necessary for maintenance of the MQC systems. Selective removal of dysfunctional mitochondria is extremely complex and requires mitochondrial and cytosolic proteins to perform highly coordinated functions to maintain healthy mitochondria; this is essential for cell metabolism and survival.23) An important pathway in mitophagy is the PTEN-induced kinase 1 (PINK1)/Parkin pathway. Under physiologic conditions, the PINK1 is imported into the mitochondrial matrix to be degraded by LonP1. However, in membrane deficient mitochondria, PINK1 accumulates on the outer mitochondrial membrane.31) This leads to the recruitment of E3 ubiquitin ligase and Parkin from the cytosol and transference to the mitochondrial membrane (PINK1-dependent Parkin translocation), where Parkin initiates ubiquitination of multiple outer membrane proteins.32) Mitophagy also takes place through a Parkin-independent pathway. Some mitophagy receptor proteins on the mitochondrial outer membrane (e.g., BNIP3, FUNDC1, Bcl2-L-13, AMBRA1, and cardiolipin) bind to LC3 in a Parkin-independent manner.33) Under hypoxic conditions, BNIP3 promotes mitophagy and protects cells from oxidative damage.34) FUNDC1 interacts with LC3 to activate Parkin-independent mitophagy in response to hypoxia and hypoxic mitophagy in platelets protecting the heart from IR injury in the FUNDC1-dependent mitophagy.35) Bcl2-L-13 induces mitochondrial fragmentation and mitophagy in HEK293 cells.36) Cardiolipin, a phospholipid stabilizing OXPHOS complex, is synthesized in the mitochondrial inner membrane and translocated to the outer membrane, where it initiates mitophagy to protect the cell from cell apoptosis under mitochondrial stress conditions.11)
MITOCHONDRIAL QUALITY CONTROL AND HEART DISEASE
UPRmt and heart disease
Nicotinamide riboside (NR) is well-known for boosting UPRmt by augmenting NAD+ pools.37) Smyrnias et al.3) reported that UPRmt boosting by NR improves heart function in the mice model of pressure overload-induced heart failure. Mice were treated with NR or a control vehicle for 3 days prior to subjecting them to transverse aortic constriction (TAC) surgery. At 1 week post TAC surgery, NR treated mice showed improved left ventricle (LV) function in the echocardiogram and reduced cardiomyocyte death in histologic analysis.3) Research from Xu et al.38) also reported a UPRmt boosting by choline-attenuated cardiac dysfunction in the rat model pressure overload-induced heart failure through the SIRT1-AMPK pathway. Aortic stenosis (AS) is the clinical model of chronic pressure overload.39) Myocardial tissue from AS patients showed that UPRmt mRNA marker levels increased in AS patients compared to control subjects. In the subgroup analysis of AS patients, UPRmt markers levels had a negative correlation with cardiomyocyte tissue death, fibrosis, and cardiac damage markers.5)
UPRmt activation also showed cardioprotective effects against heart I/R injury models. Wang et al.40) examined the role of UPRmt in perfused heart IR injury models with ATF5 knock out (KO) mouse. When the UPRmt was activated using oligomycin and doxycycline treatment, WT mice hearts exhibited significant improvement in post-IR functional recovery and infarct size. However, this protective effect was absent in ATF5 KO mice hearts. Nicotinamide mononucleotide (NMN), a NAD+ boosting agent like NR, also exhibited cardioprotective effects in the IR injury model. Nadtochiy et al.41) reported that NMN pretreatment showed significant protection against IR injury (post-IR functional recovery: NMN, 42±7% vs. vehicle, 11±3%; infarct size: NMN, 34±4% vs. vehicle, 66±4%).
Mitophagy and heart disease
Mitophagy plays a key role in the removal of damaged mitochondria in response to several stresses such as hypoxia and cytosolic Ca2+ overload.42) Previous studies report that mitophagy has an essential role in the development of heart failure. Protein levels of PINK1 in LV heart samples from end stage HF patients were decreased compared with normal controls.43) Previous research indicates that PINK1 KO mice developed cardiac hypertrophy at 2 months of age,43) and loss of PINK1 increased infarct size after I/R injury.44) Parkin KO mice accumulated abnormal mitochondria with increasing age.30) Although young Parkin KO mice had normal cardiac function, these mice were more sensitive to myocardial infarction (MI) than WT mice; moreover, they possessed a reduced mitophagy, the accumulated swollen, and dysfunctional mitochondria within the heart of the MI rat model.45) Mitophagy also showed a protective role against pressure overload induced heart failure by TAC.46)
Diabetic cardiomyopathy (DCM) is a cardiac phenotype of diabetic patients. DCM is defined as the existence of abnormal myocardial structure and performance in the absence of additional cardiac risk factors.47) Diabetes-derived mitochondrial dysfunction has an essential role in DCM development.48) The DCM-affected mitochondria portray a dysregulation of Ca2+ handling, alternations in energy metabolism, and an increased oxidative stress.49) Mitophagy activation has shown protective effects against high fat diet (HFD)-induced DCM in mice studies. A study done by Tong et al.50) in Parkin KO mice reported that mitophagy was upregulated and there were severe cardiac hypertrophy and diastolic dysfunctions after mice consumption of HFD. Enhancing PINK1/Parkin-mediated mitophagy using melatonin has been found to decrease DCM-derived dysfunctional mitochondria in mice.51) Recent studies using animal models further reveal mitophagy role in heart disease (Table 1).30)43)44)45)52)53)54)55)56)
Table 1
Genetically modified mice and their cardiac phenotypes

| Model | Mitophagy | Phenotypes | Ref |
|---|---|---|---|
| Parkin KO | Decreased | Increased sensitivity to MI and doxorubicin exposure, accumulation of dysfunctional mitochondria, and oxidative damage with age, reduced life span | 45)52)53) |
| Parkin TG | Increased | Increased life span, preserved cardiac function with aging | 52)54) |
| PINK1 KO | NA | Mitochondrial dysfunction, cardiomyopathy, increased sensitivity to I/R | 43)44) |
| BNIP3 KO | NA | Decreased apoptosis and cardiac remodeling in response to I/R | 55) |
| BNIP3 TG | NA | Increased sensitivity to MI, increased apoptosis | 55) |
| NIX KO | NA | Decreased cardiac remodeling and preserved cardiac function in response to pressure overload | 56) |
| NIX TG | NA | Ventricular dilation, reduced cardiac function | 56) |
Reproduced from “Mitophagy and heart failure.” By Shires and Gustafsson, Journal of Molecular Medicine 2015;93:253-62.30)
I/R = ischemia-reperfusion; KO = knock out; MI = myocardial infarction; NA = not assessed; I/R = ischemia-reperfusion; TG = transgenic.
NOVEL CANDIDATES FOR MITOCHONDRIAL QUALITY CONTROL IN THE HEART
Urolithin A
Numerous studies have reported the beneficial effects of phytochemicals in metabolic disease, such as metabolic syndrome and CVD.57)58) Urolithin A is the well-known gut microbiota-generated small metabolite from pomegranate fruits. Natural compounds known as ellagitannins from pomegranate juice are hydrolyzed in the gut to release ellagic acid. Colonic microflora converts ellagic acid to urolithins and enter the systemic circulation.59) Ryu et al.60) reported that urolithin A improves mitochondrial proteostasis by inducing mitophagy. Urolithin A supplementation also extended lifespan in C. elegans and improved skeletal muscle function in mice.56)
The pomegranate fruit has shown beneficial effects in CVD. Pomegranate juice improves stress-induced myocardial ischemia in patients who have coronary heart disease61) and inhibits atherosclerosis development.62) These findings suggest potential benefits of urolithin A in cardiac myocytes which many studies have revealed throughout the years. In rodents, urolithin metabolites accumulate in the myocardium after urolithin A admiration.63) Urolithin A activates mitophagy signal in cardiac myocytes and suppresses cardiac fibrosis,64) reduces cardiac tissue inflammation, and improves cardiac function in the streptozotocin-induced DCM rat model.63) Recent human clinical trials revealed that urolithin A also improves mitochondrial function in human skeletal muscle.65) These results suggest that urolithin A may improve mitochondrial and heart muscle function in CVD human patients.
Spermidine
Spermidine is a natural polyamine abundant in certain foods, such as rice bran, soybeans, aged cheese, mushrooms, and broccoli.66) This polyamine has shown beneficial effects in many diseases by improving mitochondrial function. Spermidine stimulates Ca2+ uptake in isolated mitochondria from mice liver, heart, and brain.67) In mice neuroblastoma cells, spermidine treatment protects mitochondrial damage, stabilizes mitochondrial genome and membrane potential, and reduces apoptosis due to D-galactose (gal)-induced stress.68) A combination of spermidine and exercise rescues skeletal muscle atrophy in D-gal-induced aging rats.69)
In addition, spermidine portrayed cardiovascular protective effects in previous studies. Spermidine feeding led to a 10% increase in the median lifespan of C57BL/6J mice and delayed heart failure progression in Dahl rats.70) In similar studies, spermidine reduced inflammation, induced autophagy and mitophagy, increased mitochondrial respiration in the heart, and improved heart function in aged mice.71) In a human cohort study, spermidine levels declined with aging,72) and spermidine intake showed inversed association with heart failure, acute coronary artery disease, stroke, and death due to vascular disease.73) Therefore, spermidine supplementation shows promise for CVD prevention and treatment.
CONCLUSIONS
Mitochondria have numerous roles essential for energy metabolism, cell cycle control, cell development, immune response, and cell death. Scientists regard the mitochondria as a central platform in the execution of diverse cellular events74) and see vast potential in using mitochondrial dysfunction as a therapeutic target for human diseases such as CVD. Targeting of UPRmt, mitophagy, or both, as a novel therapeutic strategy with optimistic potential in improving human heart disease. Future research focusing on the machinery regulating mitochondrial proteostasis under various stress conditions is needed to unveil the new molecular pathways of MQC.
Drugs targeting the mitochondria such as NR and spermidine have already shown good clinical activity and beneficial effects in age-related human disease.75)76) Urolithin A also showed beneficial effects in elderly skeletal muscle improvement.65) These clinical data provide good promise and excitement over novel therapeutic options against heart disease as discussed within this review. Further researches aimed at identifying new compounds enhancing mitochondrial proteostasis and long-term clinical studies are needed for the prevention of heart disease and the recovery of heart function in CVD patients.
 XML Download
XML Download