

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Abstract
Sexually transmitted infections (STIs) are caused by the spread of pathogens via sexual activity and can cause serious complications if left untreated, regardless of their symptoms. Therefore, early diagnosis of STI is important, and molecular diagnostic methods for rapid detection and monitoring are needed. In this study, we evaluated a multiplex polymerase chain reaction (PCR) kit for simultaneously detecting 13 different bacterial, fungal, and viral microorganisms that cause STIs. The kit performance was evaluated for its sensitivity, lot-to-lot variation, and interference in detecting different pathogens. Additionally, its clinical usefulness was evaluated by estimating its sensitivity and specificity for clinical samples. The limit of detection (LOD) was 0.021–50.104 copies for each pathogen. In the tests of lot-to-lot, 100% of positive samples were detected at low concentrations and negative samples all showed negative results. This result confirms that there is no the variation of lot-to-lot. In the test for interference between pathogens, the efficiency of amplification for each pathogen was not significantly reduced and no nonspecific amplification product was formed. We tested 322 vaginal swab samples using the multiplex PCR kit and confirmed that its clinical sensitivity and specificity were 100% for all pathogens. This multiplex PCR kit can be used widely for rapid diagnosis and monitoring of STIs.
Go to : 
REFERENCES
1). Muralidhar S. Molecular methods in the laboratory diagnosis of sexually transmitted infections. Indian J Sex Transm Dis AIDS. 2015; 36:9–17.

2). Black CM. Current methods of laboratory diagnosis of Chlamydia trachomatis infections. Clin Microbiol Rev. 1997; 10:160–84.
3). Cunningham SA, Mandrekar JN, Rosenblatt JE, Patel R. Rapid PCR Detection of Mycoplasma hominis, Ureaplasma urealyticum, and Ureaplasma parvum. Int J Bacteriol. 2013; 2013:168742.
4). Ferná ndez G, Martró E, Gonzá lez V, Saludes V, Bascuñ ana E, Marcó C, et al. Usefulness of a novel multiplex real-time PCR assay for the diagnosis of sexually-transmitted infections. Enferm Infecc Microbiol Clin. 2016; 34:471–6.
5). Liu H, Rodes B, Chen CY, Steiner B. New tests for syphilis: rational design of a PCR method for detection of Treponema pallidum in clinical specimens using unique regions of the DNA polymerase I gene. J Clin Microbiol. 2001; 39:1941–6.
6). Orle KA, Gates CA, Martin DH, Body BA, Weiss JB. Simultaneous PCR detection of Haemophilus ducreyi, Treponema pallidum, and herpes simplex virus types 1 and 2 from genital ulcers. J Clin Microbiol. 1996; 34:49–54.
7). Sachdev D, Patel AL, Sonkar SC, Kumari I, Saluja D. Diagnosis of Neisseria gonorrhoeae using molecular beacon. Biomed Res Int. 2015; 2015:597432.
8). Wroblewski JK, Manhart LE, Dickey KA, Hudspeth MK, Totten PA. Comparison of transcription-mediated amplification and PCR assay results for various genital specimen types for detection of Mycoplasma genitalium. J Clin Microbiol. 2006; 44:3306–12.
9). Satpathy G, Mishra AK, Tandon R, Sharma MK, Sharma A, Nayak N, et al. Evaluation of tear samples for Herpes Simplex Virus 1 (HSV) detection in suspected cases of viral keratitis using PCR assay and conventional laboratory diagnostic tools. Br J Ophthalmol. 2011; 95:415–8.
10). Van Der Pol B, Williams JA, Taylor SN, Cammarata CL, Rivers CA, Body BA, et al. Detection of Trichomonas vaginalis DNA by use of self-obtained vaginal swabs with the BD ProbeTec Qx assay on the BD Viper system. J Clin Microbiol. 2014; 52:885–9.
11). World Health Organization. Guidelines for the management of sexually transmitted infections. Geneva, Switzerland: World Health Organization;2003.
12). World Health Organization. Sexually transmitted infections (STIs). Geneva, Switzerland: World Health Organization;2013.
13). Jaschek G, Gaydos CA, Welsh LE, Quinn TC. Direct detection of Chlamydia trachomatis in urine specimens from symptomatic and asymptomatic men by using a rapid polymerase chain reaction assay. J Clin Microbiol. 1993; 31:1209–12.
14). van Der Schee C, van Belkum A, Zwijgers L, van Der Brugge E, O'Neill EL, Luijendijk A, et al. Improved diagnosis of Trichomonas vaginalis infection by PCR using vaginal swabs and urine specimens compared to diagnosis by wet mount microscopy, culture, and fluorescent staining. J Clin Microbiol. 1999; 37:4127–30.
15). Burkardt HJ. Standardization and quality control of PCR analyses. Clin Chem Lab Med. 2000; 38:87–91.
16). Elnifro EM, Ashshi AM, Cooper RJ, Klapper PE. Multiplex PCR: optimization and application in diagnostic virology. Clin Microbiol Rev. 2000; 13:559–70.
17). Burg JL, Grover CM, Pouletty P, Boothroyd JC. Direct and sensitive detection of a pathogenic protozoan, Toxoplasma gondii, by polymerase chain reaction. J Clin Microbiol. 1989; 27:1787–92.
18). Noordhoek GT, Wolters EC, de Jonge ME, van Embden JD. Detection by polymerase chain reaction of Treponema pallidum DNA in cerebrospinal fluid from neurosyphilis patients before and after antibiotic treatment. J Clin Microbiol. 1991; 29:1976–84.
19). Scoular A, Gillespie G, Carman WF. Polymerase chain reaction for diagnosis of genital herpes in a genitourinary medicine clinic. Sex Transm Infect. 2002; 78:21–5.
20). Kim JJ. Cutaneous diseases of the external genitalia. J Korean Med Assoc. 2008; 51:449–54.
21). Kim SW. Diagnosis and clinical symptoms of sexually transmitted diseases. J Korean Med Assoc. 2008; 51:875–83.
22). Flahaut M, Sanglard D, Monod M, Bille J, Rossier M. Rapid detection of Candida albicans in clinical samples by DNA amplification of common regions from C. albicans-secreted aspartic proteinase genes. J Clin Microbiol. 1998; 36:395–401.
23). Battle TJ, Golden MR, Suchland KL, Counts JM, Hughes JP, Stamm WE, et al. Evaluation of laboratory testing methods for Chlamydia trachomatis infection in the era of nucleic acid amplification. J Clin Microbiol. 2001; 39:2924–7.
24). Jaton K, Sahli R, Bille J. Development of polymerase chain reaction assays for detection of Listeria monocytogenes in clinical cerebrospinal fluid samples. J Clin Microbiol. 1992; 30:1931–6.
25). Malloy DC, Nauman RK, Paxton H. Detection of Borrelia burgdorferi using the polymerase chain reaction. J Clin Microbiol. 1990; 28:1089–93.
Go to :
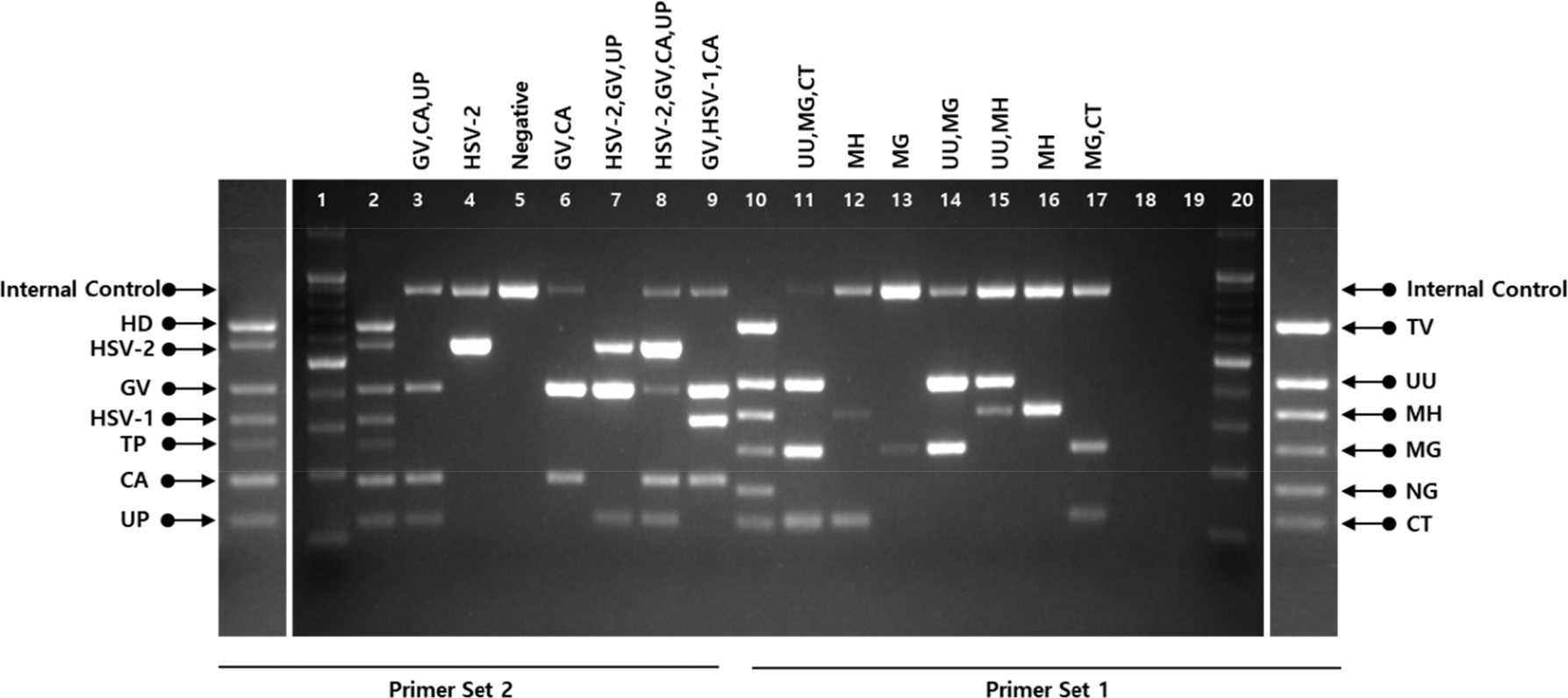 | Fig. 1.DNA amplification by multiplex PCR (E kit) in clinical specimens
The band size of each amplification product was checked against the positive control. Lanes #1 and 20: Ladder to distinguish amplification bands. Lane #2: positive control for 7 pathogens amplified by Primer Set 2, Lane #10: positive control for 6 pathogens amplified by Primer Set 1, Lanes #3-9 and #11-17: PCR results from clinical samples, Lanes #18 and 19: negative control (distilled water).
|
Table 1.
Pathogen-specific regions amplified
Abbreviations: CA, Candida albicans; CT, Chlamydia trachomatis; GV, Gardnerella vaginalis; HD, Haemophilus ducreyi; HSV-1, Herpes simplex 1; HSV-2, Herpes simplex 2; MG, Mycoplasma genitalium; MH, Mycoplasma hominis; NG, Neisseria gonorrhoeae; TP, Treponema pallidum; TV, Trichomonas vaginalis; UP, Ureaplasma parvum; UU, Ureaplasma urealyticum.
Table 2.
Limit of detection of E kit multiplex PCR
Table 3.
Lot-to-lot variation for each pathogen
Table 4.
Test for interference between CT and NG, HSV-1 and HSV-2
Table 5.
Sensitivities and specificities of E kit in 322 swab specimens
 XML Download
XML Download