

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Although there are several historical accounts of the discovery of the parathyroid glands and parathyroid hormone (PTH), Sandstrom [1] is credited with the discovery and nomenclature of the parathyroid gland, as he first came across the structure while examining a dog's neck and identified the organ in other animals in 1880. Sandstrom [1] performed human anatomical studies and named this new structure the “glandulae parathyroidae.” These findings were considered insignificant until Gley [2] rediscovered the lower parathyroid glands in rabbits and proceeded to study their function in 1891. Gley [2] observed the occurrence of tetany and death in experimentally thyroidectomized dogs only when the excised material included the glands described by Sandstrom [1]. A decade after Gley's observations, Erdheim [3] affirmed that the removal of the parathyroid glands resulted in tetany based on anatomical evidence. However, an intense debate on the role of the parathyroid glands and the cause of tetany continued for several decades. Eventually, Collip [4] resolved this controversy in 1925 by demonstrating that an acid extract of the parathyroid glands could relieve tetany after experimental parathyroidectomy. After multiple attempts and failures to isolate and obtain pure PTH from the parathyroid glands, Aurbach [5] eventually succeeded in isolating PTH using phenol extraction. Subsequently, Aurbach [5] and Rasmussen and Craig [6] separately isolated and purified PTH using countercurrent distribution methods in 1959. The amino acid sequencing of human PTH was first performed by Keutmann et al. [7] in 1978 (Fig. 1A).
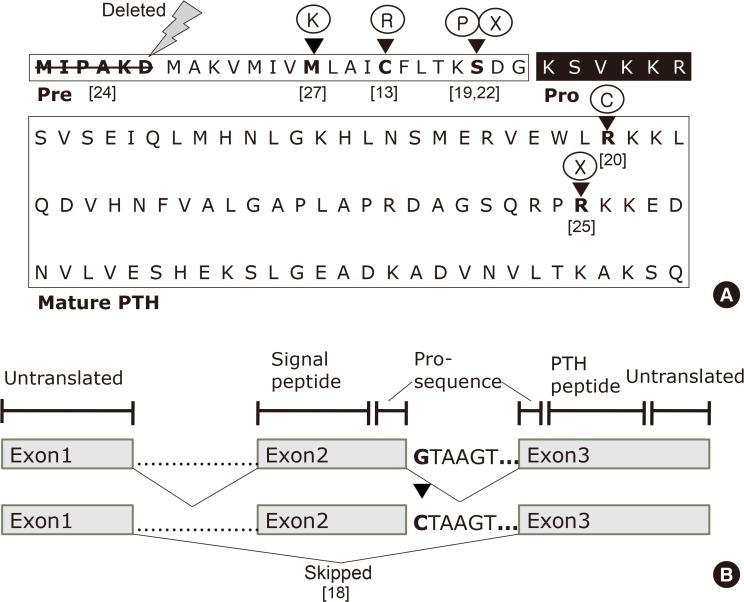 | Fig. 1(A) Primary sequence of human parathyroid hormone (PTH) and organization of its domains. The pre- (signal sequence), pro- (black box), and mature regions (boxed) of PTH are labeled. The first six amino acids in the signal sequence are deleted in a patient with hypoparathyroidism. The arrowheads indicate the wild-type amino acids in pre-pro-PTH that are replaced by mutant amino acids or terminated. (B) Schematic presentation of the human parathyroid hormone (PTH) gene, which consists of three exons and two introns. The G to C nucleotide change at the beginning of intron 2 results in exon 2 being skipped in patients with autosomal dominant familial hypoparathyroidism; [reference], see references for details [8].
|
The human PTH gene is located in the p-arm of chromosome 11 and consists of three exons and two introns (Fig. 1B) [8]. Exon 1 is an untranslated region, while the hormone is encoded by exons 2 and 3. Exon 2 encodes the signal peptide region and part of the prohormone sequence, while exon 3 is translated to form the rest of the prohormone sequence and the PTH peptide (84 amino acids), and also encodes 39 untranslated amino acids.
Isolated hypoparathyroidism refers to a group of heterogeneous conditions that result in hypocalcemia, hyperphosphatemia, and decreased bioactive PTH levels in the bloodstream. Familial isolated hypoparathyroidism (FIH) can result from several types of genetic alterations, including mutations in the calcium-sensing receptor (CASR), glial cells missing-2 (GCM2), G protein a11 (GNA11), or in the PTH gene itself [91011].
To date, eight mutations in the PTH gene have been reported in the medical literature (Table 1, Figs. 1, 2). This review presents a general overview of all the mutations detected in the PTH gene that have been reported to induce FIH and primary hyperparathyroidism.
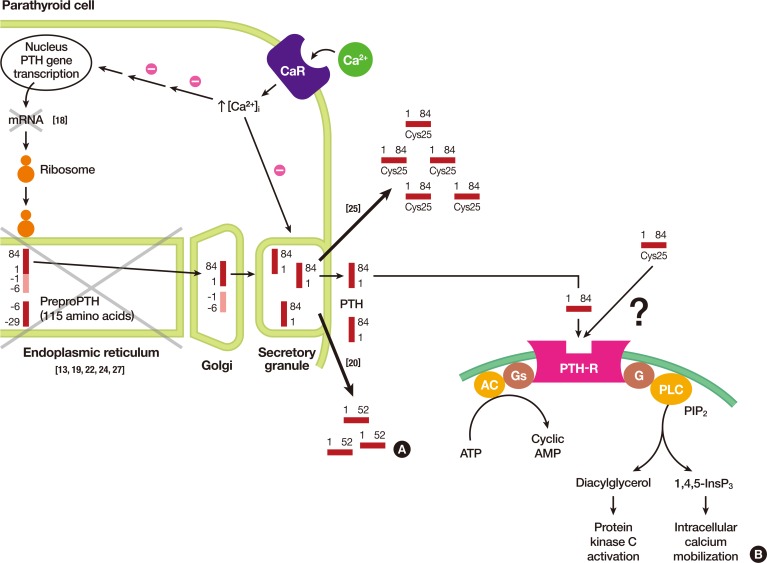 | Fig. 2(A) Illustration of parathyroid hormone (PTH) synthesis and secretion from chief cells of parathyroid glands. In each step, pathophysiological explanations are noted with x mark. (B) Signal transduction pathways from the activated PTH receptor from its respective target tissues, i.e., bone and kidney. Note that differences in signal transduction pathways activated by either wild type PTH or Cys25PTH(1-84) are still to be investigated. Adapted from Hammer et al. [17], with permission from McGraw-Hill Education. CaR, calcium-sensing receptor; AC adenylate cyclase; PTH-R, PTH receptor; G, G protein; Gs, Gs alpha subunit; PLC, phospholipase C; PIP2, phosphatidylinositol 4,5-bisphosphate; ATP, adenosine triphosphate; AMP, adenosine monophosphate.
|
Table 1
List of Patients with Reported PTH Mutations and Their Genetic Features
| Index case (sex, age) | PTH mutation | Homozygous, heterozygous, or other | Familial | Reference |
|---|---|---|---|---|
| Male, infant | c.52T>C p.C18R | Heterozygous | Yes | [13] |
| Female | c.86+1G>C exon 2 skipped | Homozygous | Yes | [18] |
| Female, 7 days old | c.67T>C p.S23P | Homozygous | Yes | [19] |
| Female, 59 years old | c.247C>T p.R83X | Somatic; hemizygous | No | [20] |
| Female, 4 months old | c.68C>A p.S23X | Homozygous | Yes | [22] |
| Female, 18 years old | c.2T>C p.M1_D6del | Heterozygous | No | [24] |
| Male, early adolescence | c.166C>T p.R56C | Homozygous | Yes | [25] |
| Male, 2 years old | c.41T>A p.M14K | Heterozygous | Yes | [27] |
![]()
Go to : 
HETEROZYGOUS PTH C18R MUTATION
In 1986, Ahn et al. [12] studied 23 affected individuals from eight families who presented with FIH and conducted a restriction endonuclease analysis in order to determine whether mutations in or close to the PTH gene could cause the autosomal disease. The data indicated an absence of gross abnormalities in the PTH gene in the affected individuals.
Four years later, Arnold et al. [13] reported a T to C point mutation in the pre-pro-PTH sequence isolated from one member of a family with an autosomal dominant inheritance pattern who participated in the previous study. This mutation changes the 18th amino acid, cysteine, to arginine and subsequently disrupts the hydrophobic core of the signal sequence by introducing a positively charged amino acid (Fig. 1A). The authors hypothesized that the mutant protein was most likely to be processed ineffectively since the hydrophobic core is crucial for efficient translocation of the secreted proteins across the endoplasmic reticulum (ER). In order to provide evidence in favor of this hypothesis and to exclude the possibility of the mutation being functionally silent, the authors constructed a pre-pro-PTH cDNA plasmid containing the aforementioned T to C point mutation and compared the in vitro translation product to pro-PTH from normal pre-pro-PTH cDNA. While both the normal and mutant pre-pro-PTH RNAs were efficiently translated, only the normal protein was effectively processed to pro-PTH. Although the authors claimed that the C18R mutation affected the interaction of the protein with the processing pathway, they could not explain the exact mechanism underlying the maintenance of the autosomal dominant inheritance pattern. They did provide a hypothetical explanation by suggesting that the mutant protein resulting from the affected allele might obstruct the pathway by successfully binding to the membrane, although it might be unable to complete the translocation process.
Four years later, Karaplis et al. [14] discovered that the C18R mutation impairs interactions between the nascent protein, signal recognition particles, and the translocation apparatus, in addition to resulting in ineffective cleavage of the mutant signal sequence by the solubilized signal peptidase. However, the hypothetical explanation proposed by the previous group was neither proven nor refuted. Furthermore, Suprasdngsin et al. [15] reported the same mutation in 2001 in a patient with sporadic idiopathic hypoparathyroidism.
The mechanism by which this mutation leads to FIH was elucidated in 2007 by Datta et al. [16]. The authors hypothesized that the mutant hormones resulting from the C18R alterations become predominantly entrapped in the ER, thereby exerting cytotoxic effects and leading to apoptosis. Their hypothesis was proven by demonstrating reduced ER stress signals and apoptosis in parathyroid cells in response to the introduction of a pharmacological chaperone (4-phenylbutyric acid) that reduced the intracellular accumulation of the mutant protein. Since retention of the cytotoxic mutant protein in the ER and subsequent cell death could be attributed to the actions of a single allele, it was concluded that FIH caused by the C18R mutation exhibits an autosomal dominant mode of inheritance (Fig. 2A) [17].
Go to :
HOMOZYGOUS PTH EXON 2 SKIPPING MUTATION
In 1992, Parkinson and Thakker [18] reported a case of FIH resulting from a consanguineous marriage. They investigated the PTH gene to detect abnormalities associated with PTH mRNA processing. The authors used polymerase chain reaction to amplify the three exons and the four intron-exon boundaries that were identified by direct double-stranded DNA sequencing. The difference between the sequences of the affected and normal individuals could be traced to a single-nucleotide, wherein a G to C substitution was observed in the first nucleotide of the second intron of the PTH gene. This transformation that changes the donor splice site sequence (GTAAGT) into a recognition site (CTAAG) for the restriction endonuclease DdeI led the authors to propose that hypoparathyroidism in this patient could have resulted from abnormal PTH mRNA splicing (Fig. 1B). Further investigation revealed that normal PTH cDNA consists of 258 base pairs, whereas the abnormal PTH cDNA contained 168 base pairs; the difference corresponding precisely to the size of exon 2, which is 90 base pairs. Furthermore, the nucleotide sequence of the PTH cDNA from the patient reveals that exon 2 had been skipped and lost, resulting in exon 1 being spliced onto exon 3. Since exon 2 contains the initiation codon (ATG), the resultant mutant allele cannot initiate the translation of PTH mRNA into the pre-pro-PTH protein and the translocation of the PTH peptide through the ER prior to secretion. Furthermore, this characteristic would only be expressed if both alleles were mutated, which explains the autosomal recessive mode of inheritance.
Go to :
HOMOZYGOUS PTH S23P MUTATION
In 1999, Sunthornthepvarakul et al. [19] reported a novel mutation that resulted in an autosomal recessive form of FIH among offspring from consanguineous parents. They discovered a T to C point mutation in the first nucleotide of the 23rd amino acid (serine) in pre-pro-PTH (Fig. 1A). The resulting change to proline prevents cleavage of the mutant pre-pro-peptide by the signal peptidase in the ER, as the residue is located in a critical position of the signal peptidase cleavage site. The intact pre-propeptide is anchored to the microsomal membrane and is eventually degraded in the rough ER (Fig. 2A).
Go to :
HETEROZYGOUS PTH R83X MUTATION
In 2008, Au et al. [20] investigated a case of a hypercalcemic patient secreting abnormally truncated PTH. The patient had a parathyroid adenoma that stained negative for PTH, after which the authors proceeded to sequence the exons. Doing so revealed a single-nucleotide polymorphism (SNP) causing a missense mutation at the 83rd residue, which codes for arginine (Fig. 1A). Notably, in 1992, Lim et al. [21] constructed a series of human pre-pro-PTH mutants with the carboxy-terminal region truncated at various points along its length to demonstrate the biological function of the carboxy-terminal region of PTH. When PTH was truncated at the 83rd amino acid, producing the same peptide as that found in the patient studied by Au et al. [20], signs of impaired translocation across the ER, cleavage of pro-PTH, and PTH secretion were noted. However, the truncated peptide was biologically active in the patient, as opposed to the conclusion that could be drawn based on the in vitro results.
Go to :
HOMOZYGOUS PTH S23X MUTATION
Thirteen years after Sunthornthepvarakul et al. [19] reported the S23P mutation, Ertl et al. [22] discovered a C to A SNP that alters the same 23rd residue, which encodes serine, to a premature stop codon (Fig. 1A). Both mutations were detected in an individual born from a consanguineous marriage and the mode of inheritance was autosomal recessive in both cases. In contrast to the observations of Sunthornthepvarakul et al. [19], the amino acid substitution present in the individual studied by Ertl et al. [22] generates a stop codon that is predicted to terminate translation at a point within the signal peptide of pre-pro-PTH, resulting in a truncated version of the protein devoid of cleavage sites for further processing.
Go to :
HETEROZYGOUS p.Met1_Asp6del MUTATION
Goswami et al. [23] conducted an extensive study on idiopathic hypoparathyroidism between 1998 and 2003 in India. No mutations or other significant findings were observed in the 49 patients with idiopathic hypoparathyroidism and the 55 controls who participated in the study; however, several SNPs were detected in pre-pro-PTH gene sequences [23]. The same group of authors extended their research in 2011 and conducted a case-control study with 147 patients and 348 controls [24], and incidentally discovered a p.Met1_Asp6 deletion due to a T to C mutation (Fig. 1A).
Go to :
HOMOZYGOUS PTH R56C MUTATION (R25C IN MATURE PTH)
In 2015, Lee et al. [25] identified a homozygous R-to-C mutation at residue 25 (R25C) of the mature PTH (1-84) polypeptide isolated from three siblings affected by FIH (Fig. 1A). Three members of this family showed hypocalcemia and hyperphosphatemia, with either considerably elevated PTH levels or with low-normal PTH levels, depending on the type of PTH assay employed. Although there were no apparent signs of a consanguineous marriage, the family's ancestors were from a remote island on the western coast of Korea and had lived there for several generations. Previous reports on various mutations causing FIH traced the mutations to the hormone's pre-pro leader segment, resulting in impaired hormone synthesis or secretion. However, this mutation is located in the mature bioactive region of PTH and does not affect hormone synthesis and secretion, as was further confirmed by an analysis of both COS-7 cells transfected with mutant PTH and blood samples from patients. With respect to the biological properties of Cys25PTH(1-34), the mutant PTH was approximately three-fold weaker than wild-type PTH(1-34) in terms of binding to the PTH 1 receptor (PTH1R) in the membrane and producing cyclic adenosine monophosphate (cAMP) in a human osteoblast-derived SaOS-2 cell line. Furthermore, although a single injection of high-dose wild-type PTH(1-34) and Cys25PTH(1-34) led to a comparable increase in plasma and urine cAMP levels in mice, and both peptides showed a similar calcemic effect, the continuous infusion of Cys25PTH(1-34) led to small but significant changes in blood calcium and phosphorus levels compared to those that resulted from wild-type PTH(1-34) infusion. Cys25PTH(1-34) was found to display a specific pattern of interaction with PTH1R, which explains the maintenance of relatively high blood calcium levels in patients that helped prevent the onset of more severe hypocalcemia. However, it remains to be determined why the parathyroid glands in these patients did not secret greater quantities of mutant PTH, such that completely normal blood calcium levels could be maintained. It was suggested that there might be some intrinsic defect in the interactions between the mutant form of PTH and PTHR1 that could not be compensated by a larger quantity of ligand (Fig. 2A). Four years later, White et al. [26] discovered that extracellular calcium prolonged the retention time of ligands on the receptor, thereby increasing the duration of receptor activation and cAMP signaling. However, this positive calcium allostery was amiss in the mutant Cys25PTH(1-34), suggesting that calcium allostery may play a substantial role in the primary signaling processes that regulate calcium homeostasis. The unique action of Cys25PTH(1-34) in cellular endosomal cAMP production and calcemic responses in patients will help determine the relevance of the association between the disease and endosomal PTHR signaling (Fig. 2B) [17].
Go to :
HETEROZYGOUS PTH M14K MUTATION
Cinque et al. [27] identified and reported a heterozygous substitution of methionine to lysine in the 14th residue of the signal peptide encoded by exon 2 of the PTH gene (Fig. 1A). An in silico analysis predicted that this mutation would impair the cleavage of pre-pro-PTH into pro-PTH. Furthermore, functional in vitro assays using HEK293 cells revealed increased intracellular retention, along with the impaired secretion of mutant peptides into the medium in M14K mutants compared to the wild type. The addition of the pharmacological chaperone 4-phenylbutyric acid reduced cellular retention and increased the accumulation of the M14K mutant in the cell medium. These findings imply that the M14K mutation might act in a manner similar to that of the C18R mutation by inducing ER stress and subsequent cellular apoptosis (Fig. 2A).
Go to :
CONCLUSIONS
Diverse and intensive molecular genetic studies of rare diseases have deepened our understanding of these diseases and have increased the likelihood of discovering new information on human physiology. This knowledge is directly applicable to the treatment of commonly associated diseases. Mutations in the PTH gene are primarily detected in rare diseases, and studies on familial hypoparathyroidism have resulted in a broader understanding of the physiological function of the parathyroid glands and a deeper molecular genetic knowledge of these glands. It is now possible to develop novel treatment methods for more common diseases, such as osteoporosis and other metabolic bone and mineral diseases, further improving our chances of applying treatment methods that were conceptualized previously.
Go to :
 XML Download
XML Download