

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
The efficacy of new cancer treatments is determined by their ability to improve overall survival (OS) in phase III randomized controlled trials (RCTs). Therefore, late-stage and pivotal trials should be based on improving OS. However, the use of OS as the primary endpoint requires a long follow-up period and a large number of patients, which translate to large trial expenses. Furthermore, OS is often influenced by confounding factors, such as more effective subsequent therapies and a crossover study design. On the contrary, image-based surrogate endpoints (IBSEs), such as progression-free survival (PFS), can provide more rapid measures of treatment efficacy and are not influenced by subsequent therapies. Developing such endpoints will also be crucial for evaluating therapies targeting small subpopulations of patients with tumors based on specific molecular aberrations (1). However, the use of IBSEs is controversial because the association between IBSEs and OS remains unknown in many tumor types. Treatment efficacy can be overestimated when the IBSEs used in a study do not correlate with OS. Therefore, the true relationship of IBSEs to OS should be validated in phase III RCTs (2).
In glioblastoma research, a trial design is particularly challenging because of the rarity of the disease and the heterogeneity related to specific genetic mutations, which are the subject of many studies on targeted therapies. IBSEs, including PFS, 6-month PFS (6moPFS), 12-month PFS (12moPFS), median PFS, and objective response rate (ORR), are gaining popularity as primary or secondary endpoints in phase III RCTs on glioblastomas (345678910111213141516171819202122232425), although the current data only support their use in screening for effective drugs in phase II trials on glioblastomas. In addition, glioblastoma is subject to the unique phenomenon of pseudoprogression, which affects response assessment (2627). Pseudoprogression in newly diagnosed glioblastoma refers to treatment-associated tumor changes that mimic progressive signs although the tumor actually remains stable or regresses on follow-up. Conversely, the term pseudoresponse refers to the phenomenon in which a tumor appears to respond to a treatment, such as an antiangiogenic agent, although it actually remains stable or progresses. These phenomena may weaken the association between IBSEs and OS.
The quality of efficacy reporting in glioblastoma phase III RCTs using IBSEs is also of major importance. The bias in efficacy reporting when using IBSEs is not negligible and can influence the reader's interpretation of the treatment efficacy, eventually harming medical decision making. To more efficiently validate promising candidate therapies for glioblastoma, we assessed the relationship between IBSEs and OS as primary or secondary endpoints for variable targeted therapies at the phase III trial level (28). Here, we determined the optimized IBSEs in targeted therapies for glioblastoma through a systematic review and meta-analysis of phase III RCTs.
MATERIALS AND METHODS
Identification of Eligible Trials
OVID-MEDLINE and EMBASE were searched to identify all published phase III RCTs on newly diagnosed or recurrent glioblastoma. The following search terms were used: ((glioblastoma) OR (GBM)) AND ((“overall survival”) OR (OS) OR (“progression free survival”) OR (“progression-free survival”) OR (PFS)) AND ((“phase 3”) OR (“phase III”)).
Trials evaluating targeted therapies for cell cycle pathways, antiangiogenic therapy, targeted therapies for growth factor receptors and their downstream pathways, or immunotherapy in at least one arm were eligible for inclusion (29). The eligible trials should have a control group receiving a placebo or standard treatment (temozolomide or bevacizumab). The search start date was not predefined, and the cutoff date was December 2017. The search was not limited by sample size, study design, selected endpoints, type of control, line of treatment, or blinding. There were also no limits on the geographic origin of the trial, language of publication, or sponsorship. Meeting abstracts without published original articles were not considered eligible for this study. Trials subsequently reported in full were excluded.
Phase III RCTs involving anaplastic astrocytomas or anaplastic oligodendrogliomas were excluded because of the distinct biological characteristics of these tumors and the different treatment planning for these diseases. Review articles, meta-analyses, systematic reviews, phase I and II trials, nonrandomized trials, observational studies, and case reports were also excluded.
Data Extraction
A standardized predesigned data extraction form was utilized. The data were independently extracted from each eligible trial by two authors, and included publication year, sample size, newly diagnosed glioblastoma versus recurrent glioblastoma, line of treatment, treatment categorization, response assessment criteria, whether the primary endpoint was defined, and other endpoints. If the primary endpoint was not explicitly reported, the primary endpoint was selected on the basis of the reported endpoints, methods described in the study objectives, statistical analysis, or sample size calculation. The presence of intent-to-treat (ITT) analysis and sponsorship were also assessed. The data retrieved from all trials were crosschecked. In cases of unclear definitions, a third author was consulted for consensus.
The IBSEs included PFS and ORR. For the definition of IBSEs, OS, which is a direct measure of clinical benefit to a patient, was defined as the time from randomization to death of any cause. PFS was defined as the time from randomization until the first evidence of tumor progression or until death of any cause, whichever comes first. The 6moPFS and 12moPFS are the PFS at 6 and 12 months after randomization, respectively. ORR was defined as the proportion of patients with reduction in tumor burden of a predefined amount. For OS and PFS, the median values, hazard ratios (HRs), confidence intervals (CIs), and p values were extracted. The 6moPFS and 12moPFS data were extracted from the articles on the basis of the reported Kaplan-Meier estimates. When unreported, 6moPFS and 12moPFS were estimated from Kaplan-Meier PFS curves using Plot Digitizer 2.6.8 (plotdigitizer.sourceforge.net).
Statistical Analysis
Data are reported as medians or means. Odds ratios (ORs) and 95% CIs were calculated for 6moPFS, 12moPFS, median PFS, and ORR. Associations between IBSEs and OS were investigated using linear regression weighted by the study sample size for the HR for OS; HR for PFS; and ORs for 6moPFS, 12moPFS, median PFS, and ORR. The strength of a correlation was determined by the standardized β coefficient and defined according to the criteria of Burnand et al. (30). A correlation coefficient of > 0.6 was considered to indicate a strong association; 0.45–0.6, substantial association; 0.3–0.45, minimal association; and 0–0.3, no association. Multiple regression analysis was conducted to address the potential effect on OS of disease entity, a specific target of test treatment, and response assessment criteria. Multiple subgroup analyses were also conducted according to disease entity, types of targeted therapies (29), and response assessment criteria (Macdonald criteria versus response assessment in neuro-oncology [RANO] criteria). For the presence of ITT analysis and sponsorship, trends over time were assessed using logistic regression for binary outcomes by comparing early publication years (2000–2011) and late publication years (2012–2017).
All statistical tests were two sided, and statistical significance was defined as p < 0.05. No corrections were made for multiple testing.
RESULTS
The search identified 204 articles. Full-text reviews of 28 potentially eligible articles were performed, and 5 articles were excluded for reporting subsets of other clinical trials (n = 3) (313233) and having no surrogate endpoint (n = 2) (3435). Finally, 23 phase III RCTs published between 2000 and 2017 were identified (Fig. 1) (345678910111213141516171819202122232425). The eligible trials included 8387 patients, and there was no overlap in these patients included in 23 phase III RCTs (Tables 1, 2).
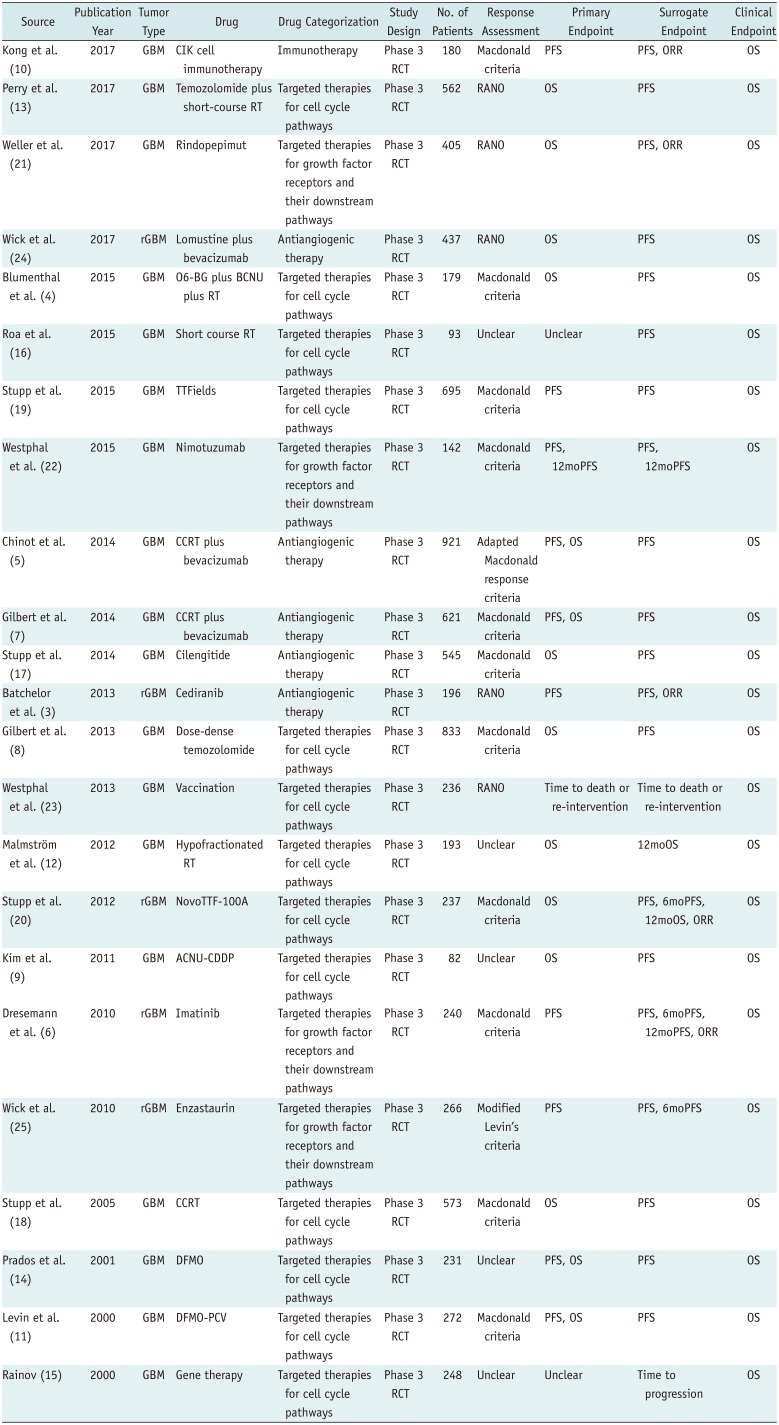
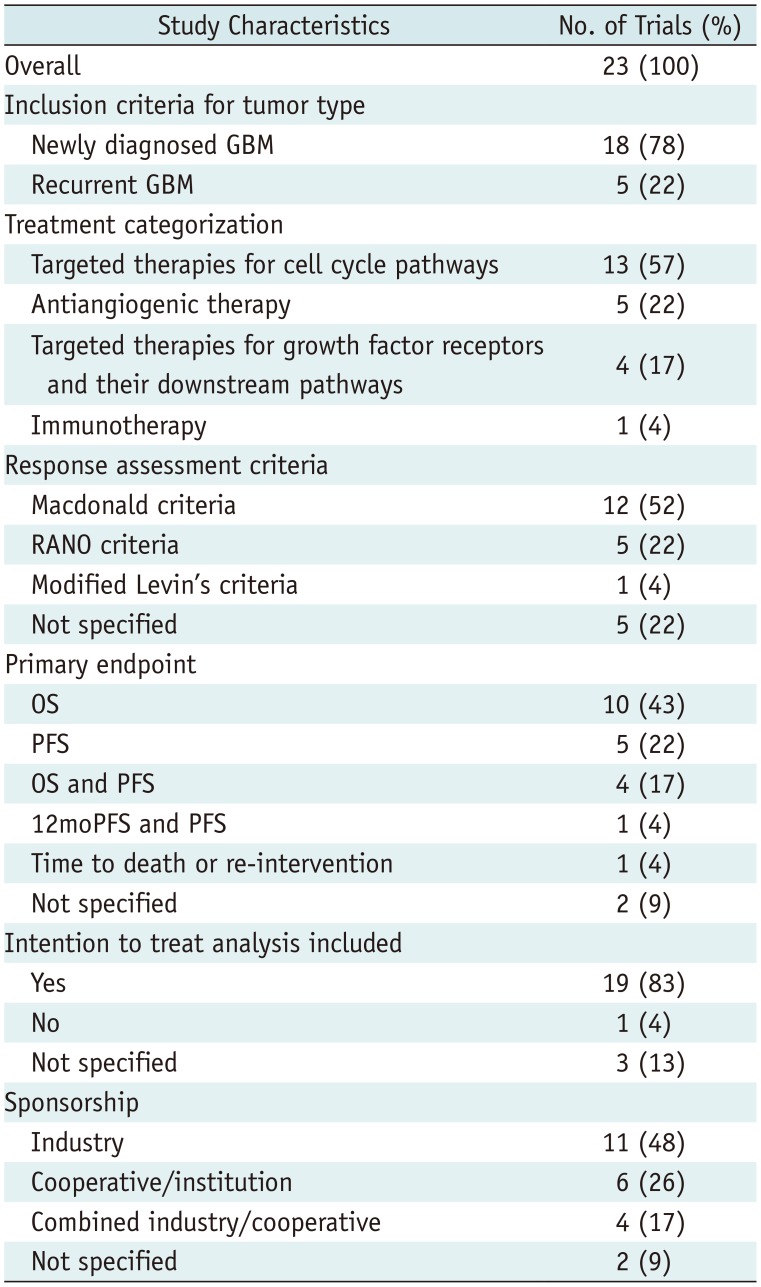
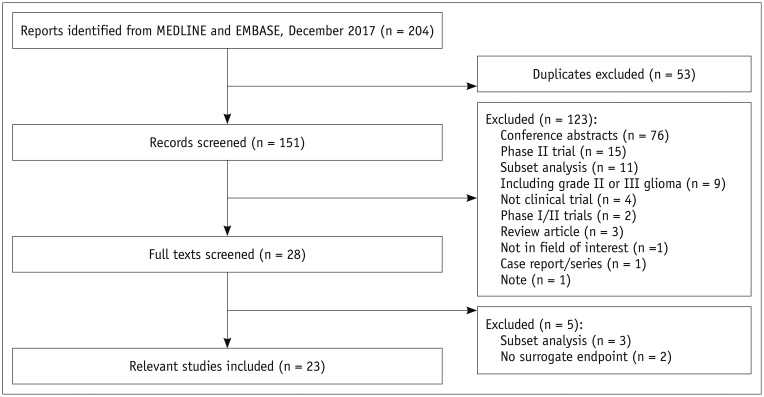
Table 1
Characteristics of Included Trials
| Source | Publicatio Year | Tumor Type | Drug | Drug Categorization | Study Design | No. of Patients | Response Assessment | Primary Endpoint | Surrogate Endpoint | Clinical Endpoint |
|---|---|---|---|---|---|---|---|---|---|---|
| Kong et al. (10) | 2017 | GBM | CIK cell immunotherapy | Immunotherapy | Phase 3 RCT | 180 | Macdonald criteria | PFS | PFS, ORR | OS |
| Perry et al. (13) | 2017 | GBM | Temozolomide plus short-course RT | Targeted therapies for cell cycle pathways | Phase 3 RCT | 562 | RANO | OS | PFS | OS |
| Weller et al. (21) | 2017 | GBM | Rindopepimut | Targeted therapies for growth factor receptors and their downstream pathways | Phase 3 RCT | 405 | RANO | OS | PFS, ORR | OS |
| Wick et al. (24) | 2017 | rGBM | Lomustine plus bevacizumab | Antiangiogenic therapy | Phase 3 RCT | 437 | RANO | OS | PFS | OS |
| Blumenthal et al. (4) | 2015 | GBM | O6-BG plus BCNU plus RT | Targeted therapies for cell cycle pathways | Phase 3 RCT | 179 | Macdonald criteria | OS | PFS | OS |
| Roa et al. (16) | 2015 | GBM | Short course RT | Targeted therapies for cell cycle pathways | Phase 3 RCT | 93 | Unclear | Unclear | PFS | OS |
| Stupp et al. (19) | 2015 | GBM | TTFields | Targeted therapies for cell cycle pathways | Phase 3 RCT | 695 | Macdonald criteria | PFS | PFS | OS |
| Westphal et al. (22) | 2015 | GBM | Nimotuzumab | Targeted therapies for growth factor receptors and their downstream pathways | Phase 3 RCT | 142 | Macdonald criteria | PFS, 12moPFS | PFS, 12moPFS | OS |
| Chinot et al. (5) | 2014 | GBM | CCRT plus bevacizumab | Antiangiogenic therapy | Phase 3 RCT | 921 | Adapted Macdonald response criteria | PFS, OS | PFS | OS |
| Gilbert et al. (7) | 2014 | GBM | CCRT plus bevacizumab | Antiangiogenic therapy | Phase 3 RCT | 621 | Macdonald criteria | PFS, OS | PFS | OS |
| Stupp et al. (17) | 2014 | GBM | Cilengitide | Antiangiogenic therapy | Phase 3 RCT | 545 | Macdonald criteria | OS | PFS | OS |
| Batchelor et al. (3) | 2013 | rGBM | Cediranib | Antiangiogenic therapy | Phase 3 RCT | 196 | RANO | PFS | PFS, ORR | OS |
| Gilbert et al. (8) | 2013 | GBM | Dose-dense temozolomide | Targeted therapies for cell cycle pathways | Phase 3 RCT | 833 | Macdonald criteria | OS | PFS | OS |
| Westphal et al. (23) | 2013 | GBM | Vaccination | Targeted therapies for cell cycle pathways | Phase 3 RCT | 236 | RANO | Time to death or re-intervention | Time to death or re-intervention | OS |
| Malmström et al. (12) | 2012 | GBM | Hypofractionated RT | Targeted therapies for cell cycle pathways | Phase 3 RCT | 193 | Unclear | OS | 12moOS | OS |
| Stupp et al. (20) | 2012 | rGBM | NovoTTF-100A | Targeted therapies for cell cycle pathways | Phase 3 RCT | 237 | Macdonald criteria | OS | PFS, 6moPFS, 12moOS, ORR | OS |
| Kim et al. (9) | 2011 | GBM | ACNU-CDDP | Targeted therapies for cell cycle pathways | Phase 3 RCT | 82 | Unclear | OS | PFS | OS |
| Dresemann et al. (6) | 2010 | rGBM | Imatinib | Targeted therapies for growth factor receptors and their downstream pathways | Phase 3 RCT | 240 | Macdonald criteria | PFS | PFS, 6moPFS, 12moPFS, ORR | OS |
| Wick et al. (25) | 2010 | rGBM | Enzastaurin | Targeted therapies for growth factor receptors and their downstream pathways | Phase 3 RCT | 266 | Modified Levin's criteria | PFS | PFS, 6moPFS | OS |
| Stupp et al. (18) | 2005 | GBM | CCRT | Targeted therapies for cell cycle pathways | Phase 3 RCT | 573 | Macdonald criteria | OS | PFS | OS |
| Prados et al. (14) | 2001 | GBM | DFMO | Targeted therapies for cell cycle pathways | Phase 3 RCT | 231 | Unclear | PFS, OS | PFS | OS |
| Levin et al. (11) | 2000 | GBM | DFMO-PCV | Targeted therapies for cell cycle pathways | Phase 3 RCT | 272 | PFS, OS | PFS | OS | |
| Rainov (15) | 2000 | GBM | Gene therapy | Targeted therapies for cell cycle pathways | Phase 3 RCT | 248 | Unclear | Unclear | Time to progression | OS |
ACNU-CDDP = nimustine plus cisplatin, BCNU = carmustine, CCRT = concurrent chemoradiotherapy, CIK = cytokine-induced killer, DFMO = difluoromethylornithine, GBM = glioblastoma, ORR = objective response rate, OS = overall survival, O6-BG = O6-benzylguanine, PCV = procarbazine, lomustine, and vincristine, PFS = progression-free survival, RANO = response assessment in neuro-oncology, RCT = randomized controlled trial, rGBM = recurrent GBM, RT = radiation therapy, TTFields = tumor-treating fields, 6moPFS = 6-month PFS, 12moOS = 12-month OS, 12moPFS = 12-month PFS
![]()
Table 2
Study Characteristics
![]()
Eighteen trials (78%) included newly diagnosed glioblastoma (4578910111213141516171819212223) and five trials included recurrent glioblastoma (36202425). Thirteen trials (57%) evaluated targeted therapies for cell cycle pathways (4891112131415161819
2023), whereas the other trials evaluated antiangiogenic therapy (n = 5) (3571724), targeted therapies for growth factor receptors and their downstream pathways (n = 4) (6212225), or immunotherapy (n = 1) (10). Twelve trials (52%) used the Macdonald criteria (36) as response assessment criteria (4567810111718192022), five trials used the RANO (37) criteria (313212324), one trial used modified Levin's criteria (25), and five trials did not report the exact response assessment criteria (912141516). The primary and secondary endpoints are described in Supplementary Materials.
Nineteen trials (83%) included ITT analysis (34568910121315161718192021232425) and one trial used a per-protocol analysis (22), whereas three trials did not explicitly report the analysis (71114). There was no statistical difference in the use of ITT-based analysis over time (OR, 0.935 [95% CI, 0.728–1.200]; p = 0.598). Sponsorship was reported in 21 trials (91%), among which 11 (48%) were sponsored by industry (3581017192021222325), 6 (26%) were sponsored by a cooperative group or an institution (479111416), and 4 received combined funding (12131824). Compared with trials published between 2000 and 2011, those published between 2012 and 2017 were more likely to be supported by industry (29% versus 81%; OR, 10.833 [95% CI, 1.374–85.440]; p = 0.024).
Association between IBSEs and OS
Our analysis of the use of surrogate endpoints only included those trials reporting the HR for OS, and either the HR for PFS or data allowing the calculation of the OR for 6moPFS, 12moPFS, median PFS, or ORR. Eighteen HR pairs for OS and PFS were available from 18 trials with 7729 patients (345678101113141718192021222425). Binary proportions for 6moPFS were reported in 12 trials and estimated from Kaplan-Meier PFS curves in 5 trials, whereas 12moPFS values were reported in 14 trials and estimated from Kaplan-Meier PFS curves in 3 trials. Binary proportions of median PFS were reported in 18 trials; however, ORR was reported in only 5 trials.
The standardized β coefficients for the weighted linear regression of OS with IBSEs are listed in Table 3. The standardized β coefficient (R) for the relationship between the HR for OS and the HR for PFS was 0.719, indicating a strong correlation between OS and PFS (Fig. 2). OS also showed a strong correlation with 6moPFS and 12moPFS; however, there was only a minimal correlation between OS and median PFS and there was no correlation between OS and ORR. The correlation between the HR for OS and the HR for PFS did not seem to be influenced by disease entity (R = −0.222, p = 0.282), types of targeted therapies (R = −0.160, p = 0.458), or response assessment criteria (R = −0.172, p = 0.404).
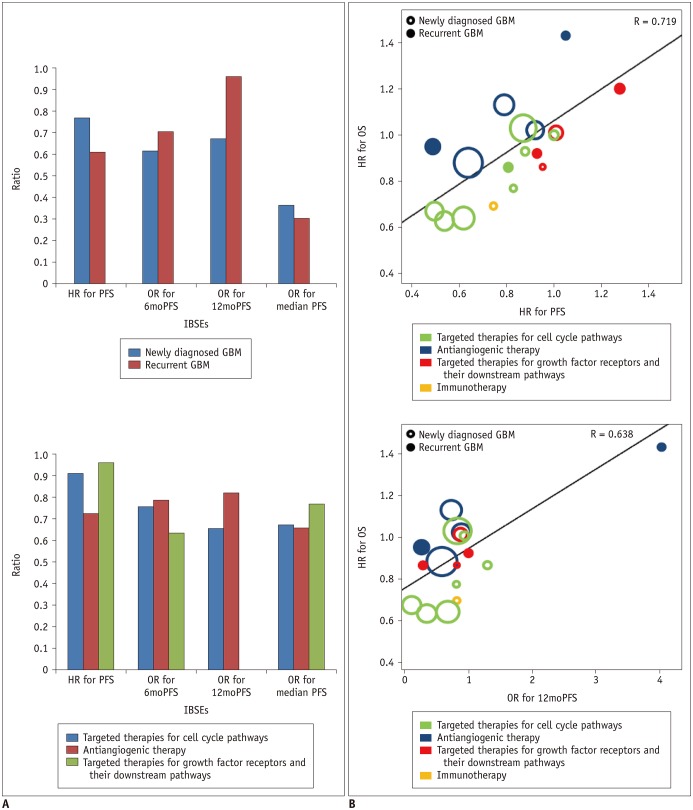
Fig. 2
Correlation of PFS with OS according to disease entity and types of targeted treatment.
A. Bar charts show correlation coefficients according to disease entity and types of targeted treatment. B. Plots show correlation of HR for PFS with HR for OS and that of OR for 12moPFS and HR for OS according to disease entity and types of targeted treatment. GBM = glioblastoma, HR = hazard ratio, IBSEs = image-based surrogate endpoints, OR = odds ratio, OS = overall survival, PFS = progression-free survival, 6moPFS = 6-month PFS, 12moPFS = 12-month PFS
![]()
Optimal Choice of IBSEs Depending on Disease Entity, Types of Targeted Therapies, and Response Assessment Criteria
In multiple subgroup analyses according to disease entity, a specific target of test treatment, and response assessment criteria, consistent strong correlations between OS and PFS were observed (R = 0.612–0.975) (Figs. 2, 3). The subgroup analysis according to disease entity revealed strong correlations between OS and 6moPFS and between OS and 12moPFS, in both newly diagnosed and recurrent glioblastoma (Table 4). However, there was only a minimal correlation between OS and median PFS in both patient groups. In recurrent glioblastoma, 12moPFS showed the highest correlation with OS (R = 0.963).
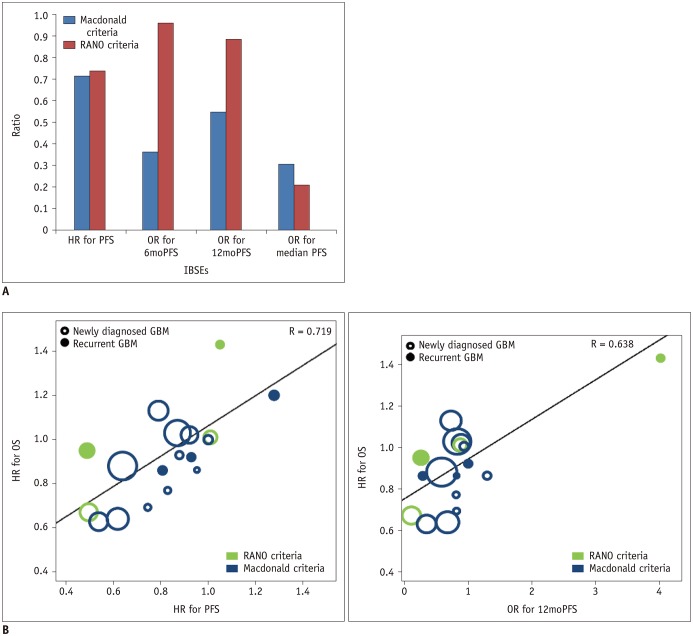
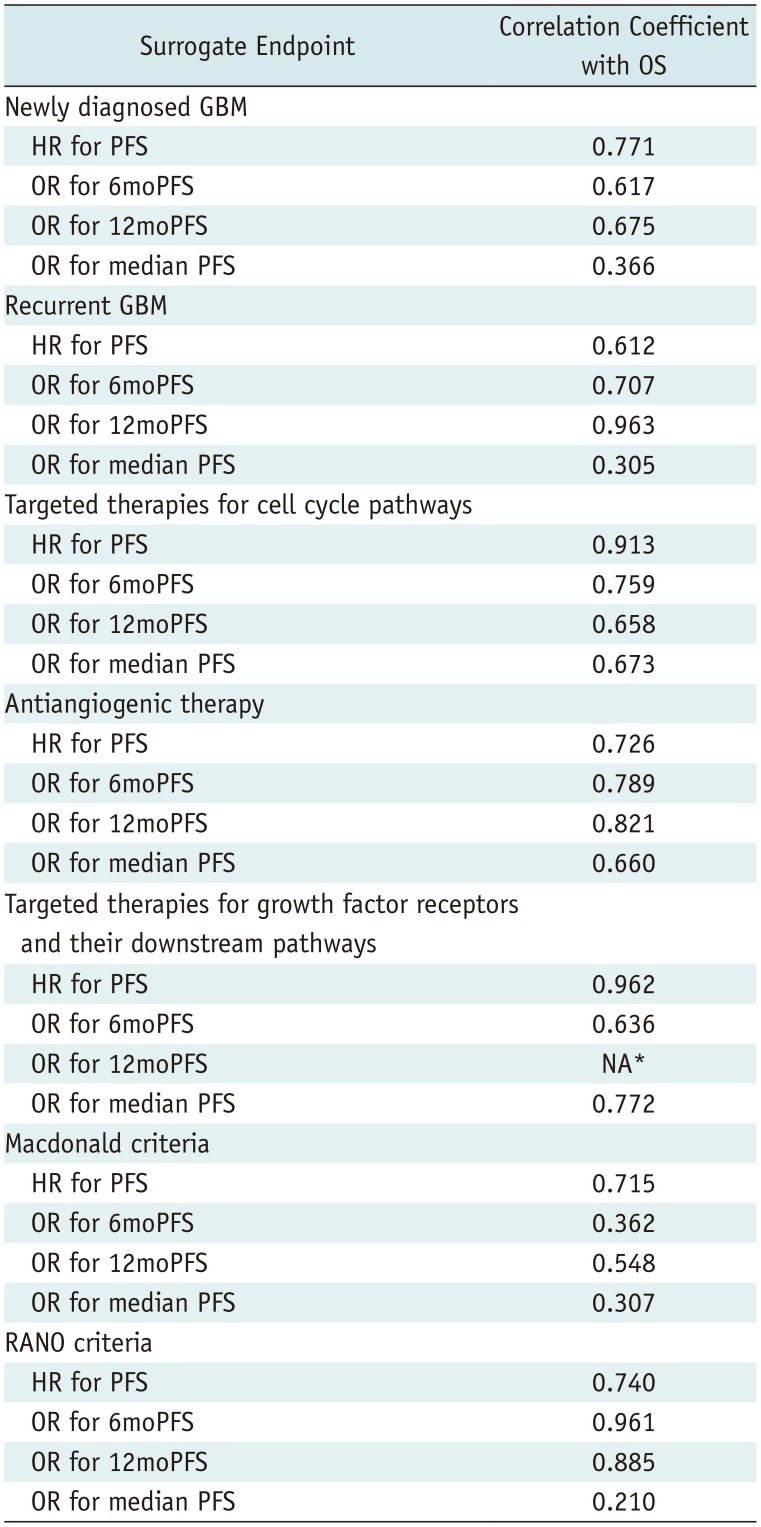
Fig. 3
Correlation of PFS with OS according to disease entity and response assessment criteria
A. Bar chart shows correlation coefficients according to response assessment criteria. B. Plots show correlation of HR for PFS with HR for OS and that of OR for 12moPFS and HR for OS according to disease entity and response assessment criteria. RANO = response assessment in neuro-oncology
![]()
Table 4
Correlation of Imaging-Based Endpoints with OS according to Subgroup Analyses
![]()
Targeted therapies for cell cycle pathways showed strong correlations between OS and 6moPFS, 12moPFS, and median PFS. PFS showed the highest correlation with OS (R = 0.913) (Fig. 2). In antiangiogenic therapy, 12moPFS showed the highest correlation with OS (R = 0.821). Targeted therapies for growth factor receptors and their downstream pathways showed strong correlations between OS and 6moPFS and between OS and median PFS, whereas there was only a minimal correlation between OS and 12moPFS. Among them, PFS showed the highest correlation with OS (R = 0.962).
The RANO criteria showed higher standardized β coefficients between OS and 6moPFS, 12moPFS, and median PFS than the Macdonald criteria (Fig. 3). Both 6moPFS and 12moPFS showed a strong correlation with OS (R = 0.961 and 0.885, respectively).
DISCUSSION
Glioblastoma is a rare tumor with heterogeneous genetic and epigenetic profiles, and with variable responses to systemic treatments. The rarity of glioblastoma is often considered a major barrier in recruiting patients to phase III RCTs, and a justification for enrolling a heterogeneous patient population with respect to genetic and epigenetic profiles. This leads to reduced statistical power in the determination of treatment benefits. In view of the difficulties in performing large phase III RCTs on glioblastoma, collecting currently available phase III RCTs on glioblastoma is important to allow research into drug activity specific to genetic mutations or epigenetic profiles.
Overall, the quality of reporting of the included trials was high. We found high proportions of clearly defined primary endpoints (91%) and ITT analyses (83%) in phase III RCTs on glioblastoma. OS, which is considered the most definitive endpoint, was used as the primary endpoint in 43% of the trials, with PFS also being used in 43%. The first phase III RCT on glioblastoma was published in the 2000, and 20 (87%) phase III RCTs were published after 2010. We believe that this may have influenced the quality of reporting compared with other studies (38). We also found that trials published between 2012 and 2017 were more likely to be supported by industry than trials published between 2000 and 2011. This is probably inevitable owing to the high costs of developing and accessing new anticancer agents in clinical trials. Similar increases in the number of sponsored clinical trials on solid tumors have been reported (3839).
To validate surrogate endpoints at a trial level, we evaluated the correlation coefficient between the treatment effects determined by PFS and OS (28). The correlation coefficient was close to 1, indicating a strong correlation between treatment effects determined by PFS and treatment effects determined by OS at the trial level. Trials in other tumor types have shown variable degrees of correlation between PFS and OS, ranging from strong correlations in soft tissue sarcoma (38) and small-cell lung cancer (40) to poor correlation in metastatic breast cancer (41). However, these studies included only a small number of trials (101112). For a successful validation of trial-level surrogate endpoints, the effective sample size (number of trials) should be sufficiently large to permit a reliable estimate of the correlation coefficient between the estimated treatment effects of the surrogate endpoints and the clinical endpoints (28). We included 18 trials reporting HR pairs for PFS and OS, and observed a strong correlation between PFS and OS with a high correlation coefficient of 0.719, which is consistent with that reported in a previous meta-analysis of phase II trials (42). Moreover, the correlation between PFS and OS did not seem to be influenced by disease entity, a specific target of test treatment, or response assessment criteria. For validation of trial-level surrogate endpoints, the HR incorporates all data from all patients and is considered to be the most appropriate measure for time-to-event outcomes (28). Therefore, these results support the use of PFS as a primary endpoint in phase III RCTs on glioblastoma.
We also observed strong correlations between 6moPFS and OS (R = 0.647) and between 12moPFS and OS (R = 0.638). However, only one trial included 12moPFS as a primary endpoint (22), and no trial included 6moPFS as a primary endpoint. A previous meta-analysis of phase II trials also showed 6moPFS to be a strong predictor of survival for recurrent high-grade gliomas (43). However, we observed only a minimal correlation between OS and median PFS. Usually, time-to-event outcomes are evaluated at a single time point (6moPFS or 12moPFS), as the median value of time-to-event outcomes (median PFS) is subject to the following problems: 1) it is likely to be affected by the choice of time point; 2) it does not consider censoring; 3) it has the possibility to be misleading if survival curves are erratic; and 4) it may not correspond well with the HR of PFS (44). There have been only five trials reporting ORR, and we found no correlation between OS and ORR. Further large-scale analyses will be needed to determine the potential correlation between ORR and OS, preferably using the RANO criteria (43). Therefore, we recommend using 6moPFS and 12moPFS as surrogate endpoints in glioblastoma trials; however, caution must be taken in using median PFS and ORR as surrogate endpoints.
The use of PFS as a surrogate endpoint offers several advantages. First, the period of time without progression is usually clinically meaningful and generally reflects treatment effects. Second, PFS is not influenced by subsequent treatments. Third, studies using PFS as a primary endpoint are usually completed in a shorter time than studies using OS as a primary endpoint. Last, but not least, studies using PFS may require a smaller sample size, as the treatment effect is often greater on PFS than on OS (45).
Recent understanding of the molecular pathways that induce glioblastoma development has led to the development of various investigational treatments specifically targeting tumor cells and the tumor microenvironment (29). Developing surrogate endpoints will be crucial for evaluating therapies targeting small subpopulations of patients with glioblastoma based on specific molecular aberrations. Random assignment in such small populations may not be feasible within the biomarker-defined group. Therefore, appropriate validation and selection of such surrogate endpoints according to a specific target of test treatment is essential to achieve success in clinical trials. Furthermore, the decision to move from early signs of efficacy based on surrogate endpoints to late-stage randomized trials is crucial because there is a high potential of overestimating efficacy by using surrogate endpoints that do not correlate with OS or through selection bias and population drift by comparison with historical controls.
In our subgroup analysis according to types of targeted therapies, PFS showed the highest correlations with OS in trials using targeted therapies for cell cycle pathways (R = 0.913) and targeted therapies for growth factor receptors and their downstream pathways (R = 0.962). In antiangiogenic therapy, 12moPFS showed the highest correlation with OS (R = 0.821). Therefore, we recommend using PFS as a surrogate endpoint in trials with targeted therapies for cell cycle pathways and targeted therapies for growth factor receptors and their downstream pathways, and using 12moPFS in trials with antiangiogenic therapy.
In the subgroup analysis according to disease entity, OS showed consistent strong correlations with PFS, 6moPFS, and 12moPFS. Particularly, PFS showed the highest correlation with OS (R = 0.771) in newly diagnosed glioblastoma, whereas 12moPFS showed the highest correlation with OS (R = 0.963) in recurrent glioblastoma. In the subgroup analysis according to response assessment criteria, trials using the RANO criteria showed higher correlation coefficients between OS and PFS, 6moPFS, and 12moPFS than trials using the Macdonald criteria. Compared with the Macdonald criteria (36), the RANO criteria included the concepts of pseudoprogression and pseudoresponse (2737). After the RANO criteria were published in 2010, trials published from 2013 started to use these criteria. We strongly recommend using 6moPFS and 12moPFS as surrogate endpoints in glioblastoma trials using the RANO criteria.
Our study had several limitations. First, in an effort to overcome the paucity of phase III RCTs on glioblastoma, we conducted an exhaustive systematic search and included all published phase III RCTs on glioblastoma, which covered a variety of different treatments and thereby included data from trials with variable sample sizes. This could have potentially increased the level of bias, although our statistical analysis was weighted for sample size. Second, the number of trials included in each subgroup, particularly for each specific target of test treatment, was low. Therefore, the results of the subgroup analyses should be interpreted with caution, and further large studies are needed.
In conclusion, the types of targeted therapies and response assessment criteria should be carefully considered for the optimal choice of IBSEs in clinical studies on glioblastoma. PFS is an optimized IBSE in targeted therapies for glioblastoma; however, 12moPFS is optimal in antiangiogenic therapy.
 XML Download
XML Download