

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Trisomy 12p is a chromosomal disorder with an estimated incidence of 1/50,000 births.1 It was first described by Uchida and Lin2 in 1973. Since then, about 50 cases have been reported to date worldwide.3
Trisomy 12p may occur both in the complete form (the entire short arm of the chromosome 12 has been copied) and in the incomplete form (only part of this short arm), as a pure form (having no other aneuploidy) or in mosaic (involving only a portion of the somatic cells of the affected individual).45 Here, we present a case of complete and pure trisomy 12p syndrome, resulting from malsegregation of a balanced translocation of maternal origin.
Case
A newborn boy was admitted to neonatal intensive care unit because of multiple fetal anomalies detected by prenatal ultrasonography. His mother was referred for evaluation of multiple congenital anomalies detected on routine sonography. This was the mother's second pregnancy and gave birth to a normal baby at the time of her first pregnancy. The ultrasonography scan performed at 35 weeks of gestation revealed a mega cisterna magna, an enlarged third ventricle, an overriding aorta, a subaortic ventricular septal defect (VSD), short femurs and short humeri. An amniocentesis was not performed. His parents were healthy and nonconsanguineous. There was no additional relevant medical history or congenital malformation. Pregnancy and delivery were uneventful.
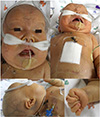
He was born at a gestational age of 37 weeks and 1 day by cesarean section. He weighed 2,560 g and appeared vigorous; the 1-minute and 5-minute Apgar scores were 8 and 9, respectively. Results of initial arterial blood gas analysis were pH 7.23, carbon dioxide pressure 62 mmHg, base excess −3.8 mmol/L, oxygen pressure 50 mmHg, and lactic acid 3.3 mEq/L. At birth, he had some dysmorphic features, including a flattened face with a broad, flat nasal bridge, a depressed forehead, upward slanting palpebral fissures, low-set and poorly lobulated ears, a long philtrum, a short neck, and clenched hands with overlapping fingers (Fig. 1). Both testes were not palpable within the scrotum. The growth profiles of birth weight, height, and head circumference were in the 10–50th percentile. Cardiorespiratory examination showed a heart rate of 140/minute and a respiratory rate of 64/minute. The blood pressure was 61/32 mmHg in the right arm and 57/29 mmHg in the right leg. On auscultation, a pansystolic murmur was heard at the left middle sternal border. Both liver and spleen were not enlarged. A 2-dimensional echocardiogram showed a large perimembranous VSD, a secundum atrial septal defect (ASD), and a large patent ductus arteriosus (PDA). Increased periventricular echogenicity was seen on the initial screening cranial ultrasonography at 2 days of age. No lesion was seen in liver, gallbladder, pancreas, spleen, both kidneys and bladder on the initial abdominal ultrasonography. Ophthalmic examination revealed no ocular disorders. Low thyroxine (T4) concentration and elevated level of 17-hydroxyprogesterone (17-OHP) were noted in the neonatal screening test. Thyroid function test confirmed free T4 0.85 ng/dL and thyroid-stimulating hormone 82.70 µIU/mL at 76 days of age and the test for 17-OHP was not repeated. The first meconium was passed 6 hours after birth. He began enteral feeding 6 hours after birth and reached full oral feeding at 5 days of age. He received a unilateral “refer” result following the automated auditory brainstem response screen, but the confirmed hearing test was not done (Table 1).
Trisomy 21 was initially diagnosed by chromosome analysis of the patient's peripheral blood, but he did not look like Down syndrome. Since fluorescence in situ hybridization using a chromosome 21-specific probe also showed negative result, chromosome analyses of his parents' peripheral blood were performed with G banding studies. After all, the complementary evaluation of the parents showed that this extra segment was the short arm of chromosome 12, which allowed for the diagnosis of complete trisomy 12p on the child [47,XY,+der(12)t(12;22) (p11.2;p13)mat] secondary to a balanced maternal translocation [46,XX,t (12;22)(p11.2;p13)] (Fig. 2).
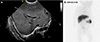
At 10 days of age, he presented with abdominal distension and radiographic pneumoperitoneum suggestive of necrotizing enterocolitis. He underwent exploratory laparotomy and small bowel resection. Exploratory laparotomy revealed a focal perforation of the distal ileum, ischemic changes of nearly the entire bowel and necrotic changes in some small bowel loops and cecum. At 13 days of age, the cranial ultrasonography conducted by a pediatric radiologist demonstrated symmetric diffuse extensive increased periventricular echogenicity and grade I intraventricular hemorrhage. Brain magnetic resonance imaging was not performed. Cholestasis was initiated at 16 days of age with direct bilirubin 2.86 mg/dL and gradually worsened until death. Abdominal ultrasonography was repeated at 23 days of age and revealed mild dilatation of right proximal intrahepatic bile ducts. At 26 days of age, both aspartate aminotransferase (AST) and Perinatologyalanine aminotransferase (ALT) rose to 545 and 298 U/L, respectively, and had increased continuously. Investigation with hepatobiliary scintigraphy at 28 days of age showed no gut excretion of technetium-99m diisopropyl iminodiacetic acid up to 24 hours suggestive of severe hepatocellular disease or biliary atresia (Fig. 3). The patient had taken ursodeoxycholic acid since 29 days of age. Percutaneous liver biopsy was considered, but not performed due to high risk of massive hemorrhage. He received mechanical ventilation until 20 days of age and started to feed again at 22 days of age and reached full feeding at 38 days of age. He underwent surgical closure of VSD at 50 days of age. AST and ALT reached their highest levels (2,020 and 1,229 U/L, respectively) at 76 days of age. The patient was stable while receiving respiratory support with heated humidified high flow nasal cannula until 76 days of age. At 77 days of age, he presented with acute respiratory insufficiency, deteriorated rapidly, and invasive ventilation was started. At 81 days of age, abdominal distension and the severity of respiratory distress worsened. The body weight of the patient measured 2 days before death was 4.2 kg (below the 3rd percentile) and the head circumference 37 cm (below the 3rd percentile). At 83 days of age, grossly bloody stools and right hemithorax were noted. Even though saline hydration, inotropic support and blood transfusion were given, he expired due to disseminated intravascular coagulation, hypovolemic shock, and multiorgan failure at 84 days of age.
Discussion
Allen et al.4 classified cases of trisomy 12p in five groups according to the extent of the 12p trisomy and the involvement of other chromosomal regions. They define “complete” trisomy as having a duplication region of 12pll or 12p12–12pter, and “pure” as having no other aneusomy or additional aneusomies of only the pter regions of non-acrocentric chromosomes, and not involving mosaicism.4 Our patient was diagnosed as a complete and pure trisomy 12p syndrome. The common physical features of the pure and complete trisomy 12p cases include normal to large birth weight, normal physical development, and severe psychomotor retardation. A prominent forehead with flat faces, hypertelorism, bilateral epicanthus, low nasal root, short upturned nose with anteverted nares, large mouth, prominent protruding lower lip, bifid uvula, micrognathia, and short neck are seen. Unlike other reports,6 full cheeks, high foreheads, and epicanthic folds were not observed from our patient and rather his face was flat. Supernumerary nipples, tapering and clinodactyly of the digits, pes planus, hallux and metatarsal valgus, and hypotonia are also near constant features, but our patient didn't have those features.
Unlike most other chromosomal abnormality syndromes, majority of trisomy 12p are born at term, with normal or above normal birth weight. An increased risk of postpartum asphyxia was previously reported.1 In our patient, the initial Apgar scores were normal but cranial ultrasonography performed on the 13th day of life revealed diffusely increased periventricular echogenicity suggesting hypoxic-ischemic encephalopathy. Neonatal complications include poor feeding and hypoglycemia.7 Our patient was fully bottle-fed and there was no episode of severe hypoglycemia.
Cardiac involvement including hypoplastic left heart was also presented in several cases of trisomy 12p, and further investigation is needed on the association with cardiac abnormalities.89101112 Despite there were no additional chromosomal imbalances other than the involvement of the short arm of chromosome 12, our patient had major congenital heart disease such as VSD, ASD and PDA.
Trisomy 12p causes problems with early developmental milestones, as well as cognitive and neurological function.7 Central nervous system abnormalities and various types of seizures have been reported in association with trisomy 12p.461314 Central nervous system abnormalities include moderate ventricular dilatation; enlargement of sylvian fissures, cortical sulci and cisterna magna; hemispheric atrophy with microgyria; internal hydrocephalus; cortical dysplasia; ectopic glial tissue in the leptomeninges; and bilateral small basal ganglia.414 Guerrini et al.15 described three cases of trisomy 12p, manifested by generalized epilepsy with myoclonic seizures or febrile seizures. Absence epilepsy was observed in another case.16 In addition, hearing loss or intellectual disability may occur.6 Since more than half of patients had some degree of hearing loss which usually developed postnatally,7 hearing evaluation is recommended. Although prenatal and postnatal cranial ultrasonography showed abnormalities, the patient in the case did not show clinically evident seizures. Most of the patients were reported to be hypotonic,6 but he had normal muscle tone.
While complete trisomy 12 is extremely rare, trisomy 12p occurs more frequently.17 The majority of reported patients with trisomy 12p resulted from malsegregation of a balanced parental translocation. Chromosomal aberration in this patient was found to be due to a balanced maternal translocation through parental examination. Trisomy 12p also can result from de novo duplication of various segments of the short arm of chromosome 12 or may also be present in only a portion of the somatic cells of the affected individual (mosaicism). The variable lengths of the trisomic segment, mosaicism, and concomitant aberrations of other chromosomes all contribute to the observed phenotypic variability of individuals with trisomy 12p.7
The patient in our case underwent surgical treatment for necrotizing enterocolitis with a focal intestinal perforation. Concomitant gastrointestinal anomalies were not noted at exploratory laparotomy. For gastrointestinal malformations, duodenal atresia, umbilical hernia and anterior anus in patients with trisomy 12p, and cardiochalasia and malrotation of the duodenum were reported in a patient with partial trisomy 12q.718 There have been no cases of hepatobiliary abnormalities related to trisomy 12 so far. Our patient developed cholestasis, but the exact cause was unknown as liver biopsy was not performed.
According to Segel et al.7 who studied the natural course of the disease, most patients had seizures after 7 years old and all patients had significant developmental delays. Duration of life of pure cases appears relatively unimpaired. But the prognosis for psycho-motor development is poor.4
Trisomy 21 was diagnosed by the initial chromosome analysis in our patient, but questions had been raised about the accuracy of the test because the appearance of the patient did not fit into Down syndrome. In the end, accurate diagnosis was possible with conducting chromosome analysis of his parents.
Besides describing clinical characteristics of a rare case of trisomy 12p, this report emphasizes the importance of the cytogenetic evaluation of the parents in cases of patients carrying structural chromosomal abnormalities. This analysis can be helpful both in clarifying the chromosomal aberration presented by the patient and in providing the appropriate genetic counseling to the family.


 XML Download
XML Download