

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Progressive Familial Intrahepatic Cholestasis type 2 (PFIC2) is an autosomal-recessive disorder caused by mutations of the ABCB11 gene which encodes the Bile Salt Export Pump (BSEP) [1]. BSEP is a bile salt transporter expressed in the canalicular membrane of the hepatocyte [23]. Its absence results in an impairment of bile salts secretion into the canaliculi leading to jaundice with severe pruritus and liver failure. The biochemical features of PFIC2 include elevated bile salts in plasma with important reduction in bile and persistently normal gamma-glutamyl transpeptidase (GGT). Histologically, it is characterized by lobular cholestasis with diffuse giant cell transformation of hepatocytes. In most cases, immunohistochemistry studies of BSEP show an absence of BSEP expression at the canalicular membrane [4]. The liver disease caused by BSEP deficiency without response to medical treatment and the increased risk of hepatic malignancy makes liver transplantation (LT) the only solution for most of these patients [25].
Although LT was classically considered curative for these BSEP patients, cholestasis recurrence with normal GGT, mediated by anti-BSEP antibodies after LT (auto-antibody Induced BSEP Deficiency, AIBD) has been recently reported [678]. These patients presented a phenotypic PFIC2 recurrence with not only biochemical but also histological features of primary BSEP deficiency [8]. It is hypothesized that patients with ABCB11 mutations causing a complete lack of BSEP expression in the native liver, derived in a lack of tolerance and anti-BSEP antibodies production after LT [7]. In severe forms, liver retransplantation is usually needed [9]. Rituximab is a chimeric monoclonal antibody against CD20, which is broadly used in hematologic malignancies and inflammatory conditions. Here we report a case of successful treatment of AIBD in an eight-year-old child using Rituximab and ten consecutive sessions of immunoadsorption. We will also highlight the potentially different clinical implications of BSEP antibodies, the possible complications related to Rituximab treatment and the need of some precautions.
This study was performed according to the guidelines of the Declaration of Helsinki and written informed consent was provided by the parents of the patient. All investigations on hepatobiliary transporters including human tissue or serum samples as presented in this study were approved by the ethical review committee of the Hospital Vall d'Hebron in Barcelona.
Go to : 
CASE REPORT
An infant girl of six months presented with severe pruritus refractory to medical treatment, a blood test showing elevated liver enzymes and bilirubin, and persistent low GGT. A mass spectrometry assay of the urine ruled out an inborn error of bile acids synthesis. At three months of age, a liver biopsy was performed showing micronodular liver cirrhosis with intrahepatic cholestasis and gigantocellular transformation. Immunochemistry for BSEP performed in liver tissue showed no BSEP expression. Genetic testing using the PFIC panel NextGeneDx (Imegen, Valencia, Spain) revealed a homozygotic pathogenic mutation (c. 1062T>A) that produces a premature stop codon [10]. The patient developed an end-stage liver disease and underwent a LT at 14 months of age. Transplant immunosuppression was induced with tacrolimus, corticoids and basiliximab. During her evolution, patient presented three episodes of biopsy proven acute rejection (BPAR) with elevated liver enzymes and GGT with normal bilirubin and bile acids (30, 54, and 70 months post-LT). All three episodes were solved after corticoids pulses and tacrolimus levels optimization. After the second BPAR, autoimmunity assessment showed high titers (1/640) of auto-antibodies against the bile canaliculi with an anti-Liver Kidney Microsoma-like pattern. Histology of the three episodes was compatible with cellular acute rejection with normal BSEP immunochemistry. In March 2017, the patient presented pruritus with cholestasis (maximum bilirubin of 14.3 mg/dL) and elevation of liver enzymes (maximum of aspartate aminotransferase 555 and alanine aminotransferase 523 UI/L) with persistent normal GGT. With the suspicion of AIBD, a liver biopsy was performed which showed intrahepatic cholestasis and giant-cell transformation with very low BSEP expression assessed by immunochemistry assay. Although none of the typical autoantibodies was observed, high titers of anti-Liver-Kidney Microsome (LKM) like antibodies were again detected. A Western Blot analysis showed that these anti-LKM like antibodies were directed against BSEP (Fig. 1) [68]. To perform the Western Blod assay, hepatocellular polarized cells from rats that expresses only a minimal level of BSEP (Can 10) [11], were transfected with a BSEPWT-green fluorescent protein (GFP) plasmid using Fugene Xtrem HP (Roche Diagnostics, Meylan, France) as previously reported [12]. Subsequently, primary antibodies used for immunostaining were incubated for 1 hour at 37°C (mouse anti-GFP; 1/80: Roche Diagnostics) and patient serum (1/100 to 1/500). Cells were rinsed and incubated with the appropriate Alexa-conjugated secondary antibodies (1/1,000; Molecular Probes, Eugene, OR, USA). The coverslips, were examined with a Zeiss Axioskop epifluorescence microscope (Carl Zeiss, Jena, Germany). Sera from patients liver transplanted for another indication than a PFIC2 were used as a negative control.
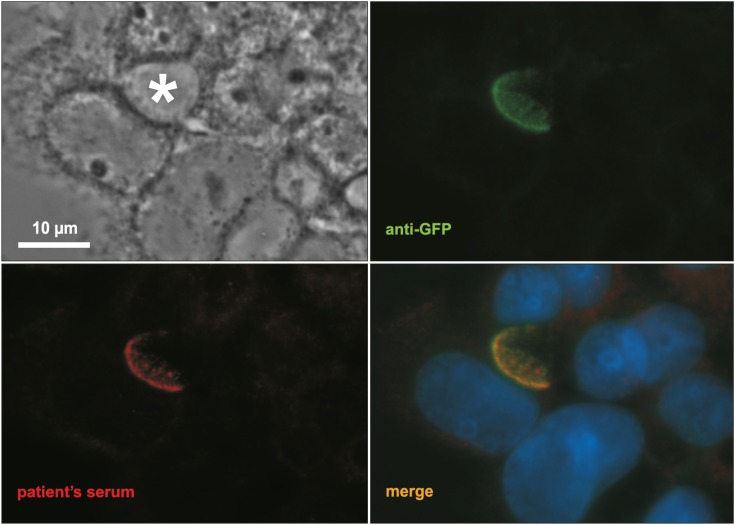 | Fig. 1Patient' serum alloantibodies are directed against BSEP. Both anti-GFP antibodies (green) and patient's serum (1/200, red) stained BSEP at the canalicular level of BSEPWT-GFP transfected Can 10 cells. * indicated a canalicular structure. Bar=10 μm.BSEP: bile salt export pump, GFP: green fluorescent protein.
|
With the diagnostic of AIBD, Rituximab at a dose of 375 mg/m2 was started and 10 sessions of immunoadsorption (TheraSorb-LIFE 18; Miltenyi Biotec, Bergisch Gladbach, Germany) were performed, decreasing the levels of liver enzymes, bile and biliary acids to their basal, nearly normal levels. The anti-BSEP titers decreased from 1/1,280 to 1/80, remaining positive despite treatment. Two months after the last Rituximab dose, the patient was admitted to our Unit because she presented dyspnoea and oxygen desaturation. Despite normal chest radiography, a CT-scan was performed observing a bilateral and diffuse pulmonary parenchymatous affectation in frosted glass pattern compatible with pneumocystis infection. In the bronchoalveolar wash pneumocystis jirovencii was detected by real-time polymerase chain reaction and direct immunofluorescence. Initially, the patient was treated using respiratory support with high flow nasal cannula, 2 mg/kg/day of corticoids and cotrimoxazol. After one week of cotrimoxazol treatment, clinical worsening with higher dyspnoea and oxygen requirements was observed. A second bronchoalveolar wash was performed that once again was only positive for pneumocystis jirovencii. Anidulafungin was added to the treatment because of its synergic effect with cotrimoxazol for treating severe pneumocystis pneumonia [13]. From there on the patient showed progressive improvement. After four weeks of cotrimoxazol and three of anidulafungin treatment, the patient was discharged from our hospital with prophylactic cotrimoxazol treatment and nearly normal liver function.
Go to :
DISCUSSION
In 2009, the first case post-LT anti-BSEP alloimmunization was reported [8]. After this first published case, less than 15 cases have been reported, evidencing it is a very rare condition. Many other genetic liver diseases need LT to be solved, but PFIC2 is the unique disease among them for which an anti-body mediated recurrence has been described. The mechanism of this recurrence is not fully understood. It is believed that severe ABCB11 mutations that carry extremely low expression of BSEP in the canaliculi of the native liver are needed to explain the lack of tolerance of the BSEP epitopes by the transplant recipient. But not all patients reported with severe mutations presented disease recurrence, possibly because BSEP-proteins act as sequestered antigens due to its intrahepatocyte localization. In some cases, these proteins can make contact with the host's immune system, especially during rejection, vascular thrombosis or ischemia-reperfusion injury. Thereby after the LT, antibodies against the normal BSEP-protein expressed by the allograft can be produced, carrying a phenotypical recurrence of the disease. In fact, BSEP recurrence can be regarded as a specific form of acute antibody mediated rejection in which the Donor Specific Antibodies are directed against BSEP. The conditions needed to develop AIBD can explain the different time lapses from LT to diseases recurrence observed between patients. In our case, it seems reasonable to think that after three BPAR episodes, BSEP epitopes were exposed, activating an antibody response. However, anti-BSEP antibodies were observed during the second episode of high GGT rejection. It is not clear why these antibodies did not initially produce liver damage or whether after the third rejection, more pathogenic anti-BSEP antibodies were produced. Although the phenotypic presentation of the BSEP recurrence is the same, it is possible that the mechanism trough which it develops is different depending on which epitope the antibody is against, therefore producing different degrees of disease. The diverse pathogenicity of the anti-BSEP antibodies could also explain the different severity observed between the reported patients. The importance of the case is mainly increase the awareness about the possibility of recurrence of the disease in PFIC2 transplanted patients. AIBD should be suspected in PFIC2 transplanted patients presenting pruritus, cholestasis and normal GGT. The suspicion should be stronger in patients with severe mutations especially after episodes of liver damage such as rejection or vascular thrombosis. The liver biopsy could reveal histological alterations similar to those found in the diagnosis, and the confirmation should be performed using Western Blot.
In some mild cases, an immunosuppression optimization is enough to decrease the production of anti-BSEP antibodies [6]. However, severe forms must be treated more aggressively in order to save the transplanted liver. Combination of immunoadsorption and Rituximab has been previously published to be effective to remove anti-BSEP antibodies [14]. In our case, two doses of weekly Rituximab and ten immunoadsorption sessions were very effective in reducing anti-BSEP titers and improving clinically and biochemically our patient. Rituximab is a monoclonal antibody that binds to CD20 antigen depleting B lymphocytes. Most of our understanding of host defense against pneumocystis has focused on T lymphocytes being almost exclusively observed in human immunodeficiency virus (HIV) patients. Although the mechanism by which Rituximab is a risk factor for pneumocystis is not known, the mortality in non-HIV Rituximab treated patients is still higher than in HIV, having more atypical presentations with a fulminant course [15]. In our patient, a rapid detection and aggressive treatment with combination therapy (cotrimoxazol plus anidulafingin) was needed to achieve infection resolution. Precaution in non-HIV immunosuppressed patients, as solid organ receptors, should be taken into account when using Rituximab, assessing case by case the need of cotrimoxazol prophylaxis.
In conclusion, AIBD should be suspected in PFIC2 transplanted patients presenting pruritus, cholestasis and normal GGT. In severe cases, combination with Rituximab and immunoadorption is an effective treatment for treating AIBD patients, but long term results are lacking. Although Rituximab is a key part of this approach, precaution against pneumocystis infection with cotrimoxazol prophylaxis should be considered.
Go to :
 XML Download
XML Download