

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Basaloid follicular hamartoma (BFH) is a rare benign tumor of the hair follicle, histopathologically characterized by anastomosing cords and strands of undifferentiated anastomosing basaloid cells1. BFH has been classified into five clinical types: (1) solitary or multiple papules, (2) a localized linear unilateral plaque, (3) localized plaque with alopecia, (4) generalized dominantly inherited familial type without an associated disorder, and (5) generalized papules associated with alopecia and myasthenia gravis2.
There were several reports of unilateral, segmentally arranged BFH with osseous, dental, and cerebral anomalies that could not be categorized into the five types of BFH. Happle and Tinschert suggested that these cases had a distinct syndrome, namely, Happle-Tinschert syndrome34. To date, only 14 cases of Happle-Tinschert syndrome have been reported. Herein, we describe another case of Happle-Tinschert syndrome.
Go to : 
CASE REPORT
A 26-year-old Korean female presented with unilateral skin lesions on the right neck, trunk, leg, and foot that had been present since birth. Her parents and brother were unaffected. She had scoliosis and her right leg was 3.5 cm shorter than her left leg. She had a history of tooth extraction due to an imbalance in the number of teeth on both sides of the jaw.
On physical examination, she had unilateral skin color change to brownish papules and patches with slight depression following the development of Blaschko's lines on the right neck, trunk, leg, and foot (Fig. 1A, B, E). Plantar pitting and linear atrophoderma with hyperpigmentation were found on the patient's right foot (Fig. 1C, D, F). In addition, onychogryphosis of the right great toenail was noted (Fig. 1C). No neurological anomalies were observed. Biopsies of a papule on the right knee and a hyperpigmented atrophic patch on the right ankle were obtained. Histopathological examination of the papule showed multiple anastomosing strands of basaloid cell proliferation with keratin material from the follicular infundibulum. Some of these cells were connected to the epidermis, and the surrounding stroma was relatively scant and loose (Fig. 2A). Histopathological examination of the specimen from the ankle showed basal hyperpigmentation and mild basaloid cell proliferation with peripheral palisading in the superficial dermis (Fig. 2B). These characteristic histopathologic features were compatible with those of BFH.
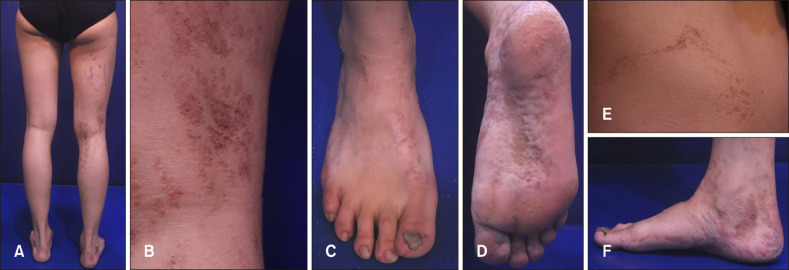 | Fig. 1Clinical features of the patient. (A, B, E) Unilateral skin color is changed to brownish papules and depressed patches following the development of Blaschko's lines on the right flank and leg. (C) Onychogryphosis of the right great toenail. (D, F) Plantar pitting and linear atrophoderma with hyperpigmentation on the right foot.
|
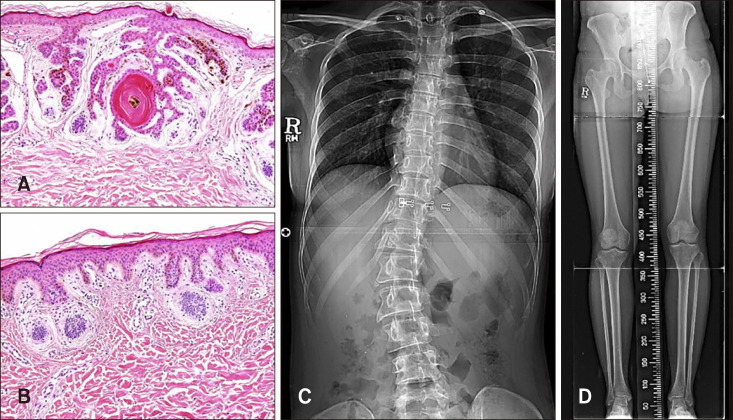 | Fig. 2Histologic features of the skin biopsies and radiographic results. (A) Multiple anastomosing strands of basaloid cell proliferation with keratin material. Some cells are connected to the epidermis with basaloid cell proliferation and scanty, loose stroma (H&E, ×200). (B) Basal hyperpigmentation and mild basaloid cell proliferation with peripheral palisading in the superficial dermis (H&E, ×200). (C, D) Radiograph showing scoliosis with a Cobb angle of 37° and shortening of the right leg.
|
The radiograph confirmed scoliosis, with a Cobb angle of 37° and shortening of the right leg (Fig. 2C, D).
To identify causative genes, we investigated genomic DNA extracted from a peripheral blood sample obtained from the patient using next-generation sequencing (NGS). NGS analysis could not identify mutations in the Sonic hedgehog pathway genes including the patched-1 (PTCH1). The smoothened (SMO) was not examined.
On the basis of the cutaneous features including unilateral, segmentally arranged, multiple BFHs with the clinical presentation of skin color change to brownish, grouped papules and atrophic patches following the development of Blaschko's line, plantar pitting, and onychogryphosis with skeletal anomalies (including scoliosis and shortening of the right leg) and dental anomalies, the patient was diagnosed with Happle-Tinschert syndrome.
We received the patient's consent form about publishing all photographic materials.
Go to :
DISCUSSION
Happle-Tinschert syndrome was first proposed in 20083, and it manifests as unilateral multiple BFHs, atrophoderma, and plantar pits with dental and osseous anomalies45678. The clinical features of Happle-Tinschert syndrome are characterized by the segmentally arranged BFHs and the presence of three systemic features: ipsilateral osseous, dental, and cerebral abnormalities (Table 1).
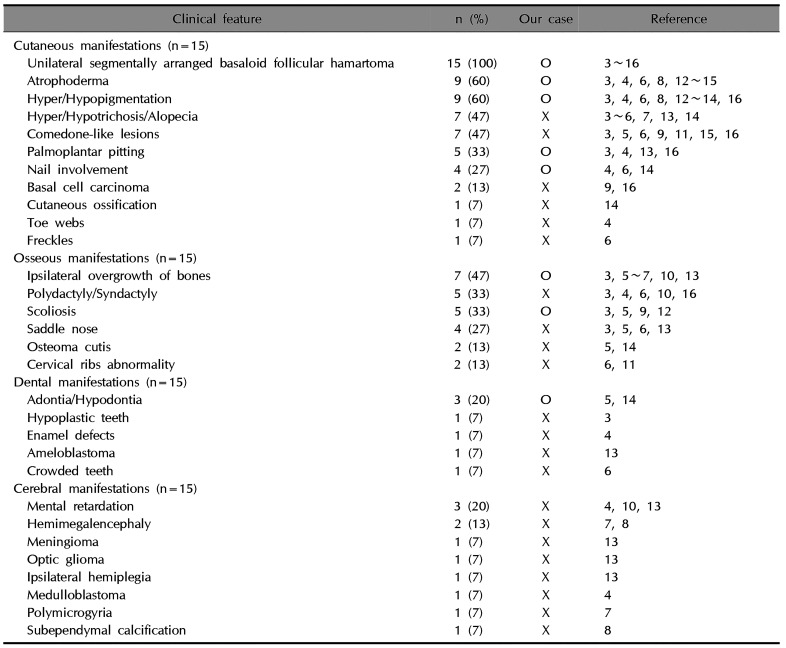
Table 1
Clinical features of Happle-Tinschert syndrome
![]()
In cutaneous manifestations, all patients presented with unilateral, segmentally arranged BFHs, and about half of them presented with comedone-like lesions, atrophoderma, hair abnormalities, and hyper/hypopigmentation345678910111213141516. Osseous manifestations, which occurred in 11 cases, were the second most common, with ipsilateral overgrowth of bones, polydactyly, syndactyly, saddle nose, and scoliosis being predominant3456791012131416. The following dental manifestations occurred in 6 cases: adontia, hypodontia, hypoplastic teeth, enamel defects, ameloblastoma, and crowded teeth34561315. Cerebral manifestations occurred in 5 cases4781013. Other accompanying anomalies are listed in Table 2. In our case, the patient showed cutaneous and osseous anomalies including unilateral, segmentally arranged BFHs followed by the development of Blaschko's line, plantar pitting, atrophoderma, dystrophic nails, hypopigmentation and hyperpigmentation, scoliosis, and shortening of the right leg. She had a history of imbalance in the number of teeth on both sides of the jaw. She did not have cerebral anomalies.
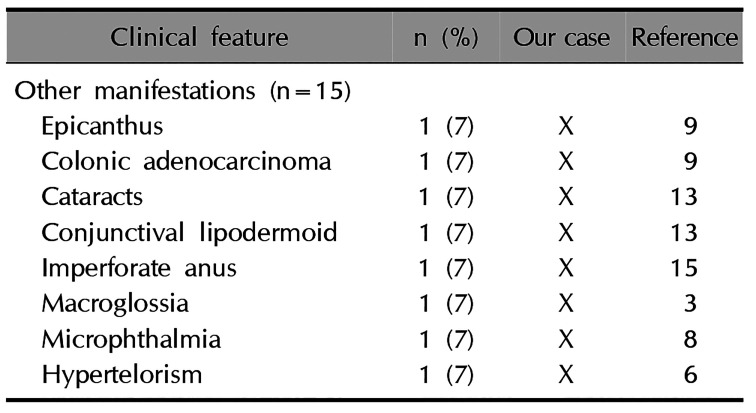
Table 2
Other accompanying symptoms
![]()
On the basis of the identification of mutations in PTCH1, which encodes the Hedgehog receptor, the pathogenesis of BFH is considered to be associated with deregulation of the PTCH signaling pathway in some familial forms of BFH. However, the pathogenesis of Happle-Tinschert syndrome is still not fully understood. Mosaic SMO mutations, encoding SMO that transduces hedgehog signaling, were revealed in 1 case17. In 2016, Khamaysi et al.18 reported a case of a heterozygous SMO mutation in Gorlin syndrome with shortening of the left first and third digits. Happle and Tinschert19 argued that the diagnosis of their patient was Happle-Tinschert syndrome, but Khamaysi et al.20 disagreed. Happle and Tinschert19 also suggested that Happle-Tinschert syndrome may be a variant of Curry-Jones syndrome, another multisystem disorder characterized by craniosynostosis, syndactyly/polydactyly, and cutaneous and intestinal abnormalities, on the basis of the finding that a mosaic mutation in SMO is the main cause of Curry-Jones syndrome and its clinical similarities. Nevertheless, further clinical and genetic studies are needed to prove this suggestion. Happle-Tinschert syndrome is not well established; thus, more consensus on its diagnosis is needed. To better understand the molecular basis and pathophysiology of this mosaic disease, identification of the mutations in DNA from the lesional tissues should be further studied.
In our case, NGS using the patient's genomic DNA from a blood sample did not show any pathogenic mutation in the Sonic hedgehog pathway genes, including PTCH1. However, there are some limitations to our genetic study: (1) our NGS panel did not include the SMO and (2) because the presumed cause of this syndrome is postzygotic mosaicism, gene analysis should have been performed using tissue obtained from the patient's skin lesion.
In conclusion, this case illustrates the characteristic clinical features of Happle-Tinschert syndrome and we report another case of this syndrome.
Go to :
 XML Download
XML Download