

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Profilaggrin, filaggrin (filament-aggregating protein), and their proteolyzed amino acids are multifunctional protein components contributing to formation of the intact stratum corneum barrier1. According to the “outside–inside” view, atopic dermatitis (AD) is a common chronic skin disease that is characterized by abnormal permeability barrier owing to the deficiency of filaggrin2. Since two prevalent loss-of-function (LOF) mutations (p.R501X and c.2282del4) of the filaggrin gene (FLG) were identified in the European populations with ichthyosis vulgaris (IV) and atopic eczema34, a comprehensive analytical strategy for screening of FLG variants has been implemented in the cohorts with not only IV or AD but also secondary allergic diseases5. Although various FLG-null mutations have been revealed in the selected Asian populations (including Japanese67, Chinese89, Singaporean Chinese10, Taiwanese11, and Korean1213), the prevalence and mutation rates of FLG-null alleles are different from European cohorts with IV or AD. Additionally, FLG LOF variants were found to be less common in patients of African ancestry with AD compared with European and Asian patients1415. A recent study revealed FLG mutations in 22.2% of African Americans with IV/AD16, whereas no FLG LOF mutations were detected in South African amaXhosa patients with AD17. Besides the genetic background predisposing to skin barrier dysfunction, non-genetic alterations of FLG seem to be correlated with AD pathogenesis in non-European populations, which have shown relatively low frequencies of FLG-null mutations.
An epigenetic trait is defined as a stably heritable phenotype resulting from changes in a chromosome without alterations in the DNA sequence18. Epigenetic mechanisms including DNA methylation, histone modifications, and non-coding RNAs have been implicated in a huge variety of human diseases correlating with gene–environment interactions1920. Methylation of the cytosine base within a CpG dinucleotide context in gene promoters is generally associated with transcriptional repression2122. Aberrant DNA methylation and subsequent silencing of a tumor suppressor gene have also been demonstrated in skin tumors, such as basal cell carcinoma, and in autoimmunityrelated skin disorders23. Recently, epigenetic alterations have been proposed as an alternative mechanism of predisposition to AD in people with wild-type FLG. Ziyab et al.24, for example, reported that DNA methylation at the single CpG site in the FLG gene body showed a significant interaction with FLG LOF variants on the risk for eczema. Nonetheless, epigenetic factors that may account for the deficiency of filaggrin and its degradation products in AD remain elusive. We hypothesized that epigenetic changes at the FLG gene promoter might contribute to modulation of FLG expression in human epidermal keratinocytes and might be associated with one of the etiopathogenic mechanisms that trigger the deficit of filaggrin in the affected epidermis of AD patients.
MATERIALS AND METHODS
Ethics statement
This study was conducted according to the Declaration of Helsinki Principles and was approved by the Institutional Review Board of the Chung-Ang University Hospital (IRB no. C2015258 [1716]). All patients gave their written informed consent to participate in the study.
Cell culture and drug treatment
Normal human epidermal keratinocyte (NHEK) cells were obtained from the PromoCell (Heidelberg, Germany). NHEK cells were grown in Keratinocyte Growth Medium 2 (PromoCell) supplemented with Keratinocyte Growth Medium 2 SupplementMix (PromoCell). All cells were maintained at 37℃ in a humidified incubator at 5% CO2. We treated the undifferentiated NHEK cells with 2′-deoxy-5-azacytidine (DAC) for 72 hours and with trichostatin A (TSA) for 24 hours. DAC and TSA were obtained from Sigma-Aldrich (St. Louis, MO, USA).
Tissue samples
Punch biopsy specimens were collected from the paired non-lesional and lesional skin of patients with severe AD (n=10). Epidermal samples were isolated under a dissecting microscope. Genomic DNA extraction was performed using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany).
Quantitative polymerase chain reaction
As described in detail previously25, total RNA was isolated from the cell lines using the RNeasy Mini Kit (Qiagen) following the manufacturer's instructions. cDNA synthesis for real-time two-step reverse transcription polymerase chain reaction (PCR) was performed on 1 µg of total RNA using the QuantiTect Reverse Transcription Kit (Qiagen), and then 1 µl of diluted cDNA was utilized for cycling reactions using the Rotor-Gene SYBR Green PCR Kit (Qiagen). The amplification and quantitative analysis were performed on the Rotor-Gene Q 5plex HRM system (Qiagen). Thermal cycling was conducted using the default conditions of the Rotor-Gene Q Series Software (Qiagen), which consisted of 5 minutes at 95℃ followed by 40 rounds of 5 seconds at 95℃ and 10 seconds at 60℃. The transcript level measured was normalized to the results of a QuantiTect Primer Assay (Qiagen) for ACTB (encoding β-actin), which served as an internal control.
In vitro methylated reporter assay
As described in detail previously25, a 928-bp fragment harboring four CpG sites located at the 5′-end region of FLG (NC_ 000001.11), a 5′ NsiI site, and a 3′ HindIII site was synthesized and cloned into the pCpGfree-basic-Lucia reporter plasmid (InvivoGen, San Diego, CA, USA) that codes for a secreted coelenterazine-utilizing variant of luciferase. Briefly, 5 µg of the Lucia reporter plasmid was methylated using 12 U of M.SssI CpG methyltransferase (New England BioLabs, Ipswich, MA, USA) in vitro at 37℃ for 20 hours. After purification with the DNA Clean & Concentrato™-5 Kit (Zymo Research, Irvine, CA, USA), methylation of the plasmid was verified by bisulfite pyrosequencing of the selected CpG units in the FLG promoter (Supplementary Fig. 1). Next, HEK293T cells were transiently transfected with unmethylated or methylated reporter plasmids by means of the jetPEI reagent (Polyplustransfection, New York, NY, USA). Luciferase activity of the Lucia reporter in the supernatant of the cells at 72 hours after the transfection was measured using the QUANTI-Luc assay reagent (InvivoGen). In each experiment, the cells were cotransfected with the pCMV-CLuc 2 Control Plasmid (New England BioLabs), which encodes a secreted variant of Cypridina luciferase, as a normalization control. Luminescence induced by Cypridina luciferase in the cell supernatant was determined using the BioLux Cypridina Luciferase Assay Kit (New England BioLabs). Reporter activity was normalized by calculating the ratio of Lucia to CLuc activities.
DNA methylation analyses
For bisulfite genomic sequencing analysis, as described in detail previously25, genomic DNA was extracted from the cell lines using the QIAamp DNA Mini Kit. Bisulfite modification of the genomic DNA was performed using the EZ DNA Methylation-Lightning™ Kit (Zymo Research). The bisulfite-converted genomic DNA was amplified using primer sets specific to the proximal promoter region of FLG (positions −738 to −162 upstream of the transcription start site (TSS), which is denoted as +1 in Supplementary Fig. 2) containing four CpG units. The sequences of the PCR primers used were as follows: forward (5′-AGAAGGAAGAGTATGTGGAATATG-3′) and reverse (5′- CAACAACCTATATTTACTTCCCAAC-3′). Cycling conditions were as follows: initial denaturation at 94℃ for 10 minutes; 45~50 cycles of denaturation at 94℃ for 30 seconds, annealing at 56℃ for 30 seconds, and extension at 72℃ for 30 seconds; and the final extension at 72℃ for 10 minutes. PCR products were purified and subcloned into the pGEM-T Easy Vector (Promega, Madison, WI, USA) for subsequent sequencing analysis. The nucleotide sequences of 12~15 independent clones were analyzed.
Furthermore, the bisulfite-modified genomic DNA obtained from normal individuals and AD patients was amplified using primer sets specific to two key CpG sites within the FLG promoter. The sequences of the PCR primers used were as follows: CpG 4 site, forward (5′-GAAGGAAGAGTATGTGGAATATGT-3′), 5′-biotinylated reverse (5′-CACTAAAAACATAAATTTAATTAAACAAAACTC-3′), and sequencing (5′- TGTGGAATATGTTTTTGATGTTA-3′); CpG 2 site, forward (5′-AGATGGAATATATAGATTAAAAAAGAATA-3′), 5′-biotinylated reverse (5′-TAAATACACTTCCTAATCCTTATCTCT-3′), and sequencing (5′-TTTTTATTATAAATTGAATTTTAAG-3′). Pyrosequencing reactions were performed using the PyroMark Gold Q96 Reagents (Qia gen) and quantitative analysis was conducted on the PyroMark Q96 ID platform (Qiagen).
Statistical analysis
Data were presented as mean±standard error of the mean and analyzed by Student's t-test. To determine significant differences in the quantitative methylation data generated by pyrosequencing measurements, comparisons between nonlesional and lesional tissue samples were made using the Wilcoxon signed-rank test. Differences with a p<0.05 were considered statistically significant. Statistical analyses were conducted using the IBM SPSS Statistics ver. 20 software (IBM Corp., Armonk, NY, USA).
RESULTS
DNA methylation-dependent regulation of FLG expression in NHEK cells
To test whether epigenetic regulation of FLG takes place in NHEK cells, we first examined the restoration of FLG expression in undifferentiated NHEK cells after treatment with either the DNA methyltransferase inhibitor DAC or the histone deacetylase inhibitor TSA (Fig. 1A). The DAC-induced DNA demethylation and TSA-mediated histone acetylation triggered a significant increase in FLG mRNA levels in undifferentiated NHEK cells. These data implied that the loss of FLG expression in undifferentiated NHEK cells correlated with epigenetic silencing involving both DNA methylation and histone modifications.
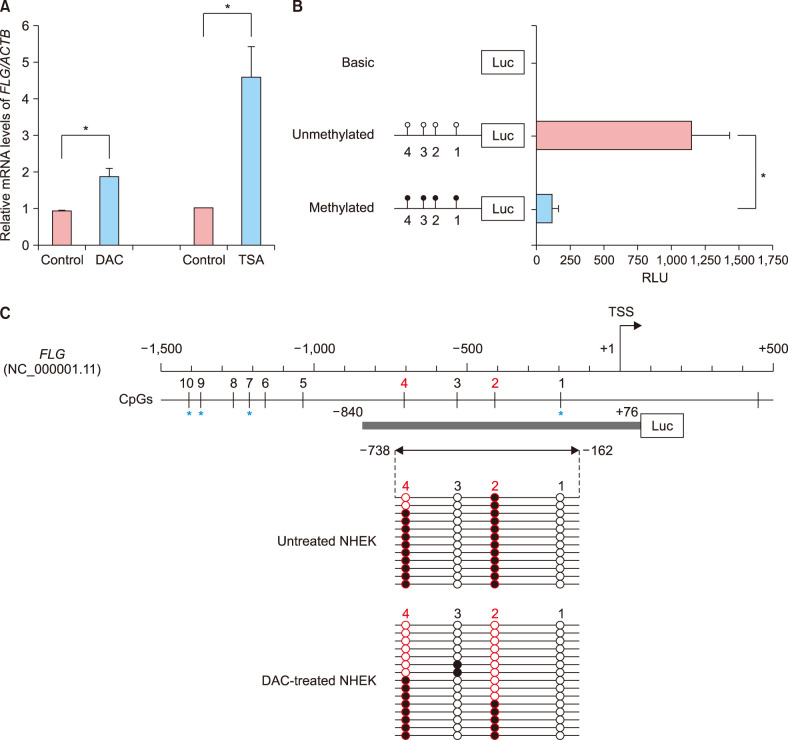
Fig. 1
Epigenetic regulation of filaggrin gene (FLG) expression in undifferentiated normal human epidermal keratinocyte (NHEK) cells. (A) Quantitative polymerase chain reaction was performed to determine the restoration of FLG mRNA expression in undifferentiated NHEK cells after treatment with DNA demethylating agent 2′-deoxy-5-azacytidine (DAC) or histone deacetylase inhibitor trichostatin A (TSA). (B) A promoter reporter assay of the 5′-end region of FLG in HEK293T cells using the pCpGfree-basicLucia reporter plasmid. The synthesized fragment contained four CpG dinucleotide sites (represented by lollipops). (C) Bisulfite sequencing analysis of the FLG promoter harboring CpG 1 through CpG 4 sites in undifferentiated NHEK cells. Circles represent stand-alone CpG sites, and each row represents the DNA sequence of an individual clone. Unmethylated and methylated CpG units are depicted as white and black circles, respectively. The blue asterisk indicates the location of CpG ID in the Illumina HumanMethylation450 BeadChip: CpG 1, cg19855573; CpG 7, cg13447818; CpG 9, cg10500702; CpG 10, cg26390526. ACTB: beta-Actin, RLU: relative light unit, TSS: transcription start site. *p<0.05.
![]()
Next, to determine whether DNA methylation in the 5′-end region of FLG was important for modulating the transcriptional activity of the core promoter, we conducted an in vitro methylated reporter assay (Fig. 1B). We synthesized an FLG fragment containing the non-CpG island (CGI) promoter and four CpG dinucleotide units therein (Fig. 1C). In vitro methylation was performed after cloning this fragment into a CpG-free luciferase plasmid to assess the role of CpG methylation in FLG transcription. HEK293T cells transfected with the methylated FLG reporter plasmid showed markedly decreased luciferase activity as compared with the cells transfected with the unmethylated FLG plasmid. The magnitude of the reduction between unmethylated control and methylated plasmid was approximately 90%, suggesting that DNA methylation directly led to transcriptional silencing of FLG with non-CGI promoter.
To identify the critical CpG sites important for regulating FLG expression in undifferentiated NHEK cells, we performed bisulfite genomic sequencing of the proximal promoter region of FLG. We determined the methylation profiles of the four CpG dinucleotides (i.e., CpG 1 to 4) located in the FLG promoter, assigning the first CpG dinucleotide to a site 185 bp upstream of FLG TSS (CpG 1). As shown in Fig. 1C, the methylation profile of the FLG promoter revealed a substantial difference in methylation frequency at both the CpG 2 and CpG 4 loci (−410 and −702 bp upstream of the FLG TSS, respectively) in the DAC-treated NHEK cells, when compared to control cells. These findings indicate that the site-specific CpG dinucleotides within the FLG promoter might be preferentially affected by a DNA demethylating agent in undifferentiated NHEK cells.
CpG methylation frequencies in the FLG promoter were higher in lesional tissues than in non-lesional tissues of AD subjects
To evaluate whether the epigenotyping for FLG by using of the putative DNA methylation marker (i.e., CpG 2 and CpG 4 dinucleotide units within the FLG promoter) was applicable to the epidermal tissues from AD patients, we performed a small-scale pilot study comparing methylation differences of these CpG loci between non-lesional and lesional epidermis obtained from Korean patients with severe AD (Eczema Area Severity Index>15; n=10; Table 1). After the triplicate experiments of bisulfite pyrosequencing for each pairs of matched samples, we observed that CpG 2 sites were characterized by overall higher methylation frequencies in lesional tissues than in matched nonlesional tissues (Fig. 2A). Although the differential methylation status at the CpG 4 unit was detected during the bisulfite genomic sequencing of FLG in undifferentiated NHEK cells, no significant correlation was observed in the quantitative methylation analysis between nonlesional and lesional epidermal tissue samples of AD subjects (Fig. 2B). Nonetheless, a statistically significant association of methylation frequencies was shown by a single CpG unit, i.e., CpG 2, within the FLG promoter (p=0.0343) in the skin biopsy samples categorized into non-lesional and lesional samples from the AD patients (Fig. 2B).
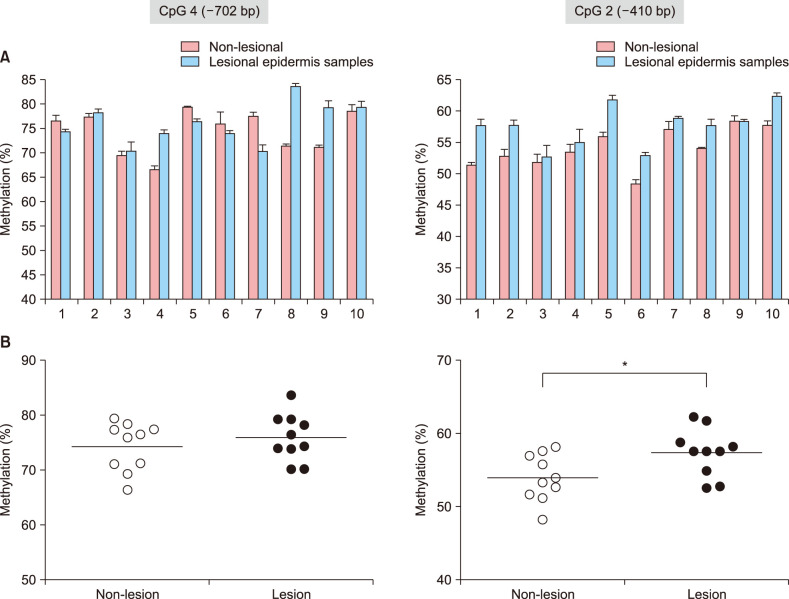
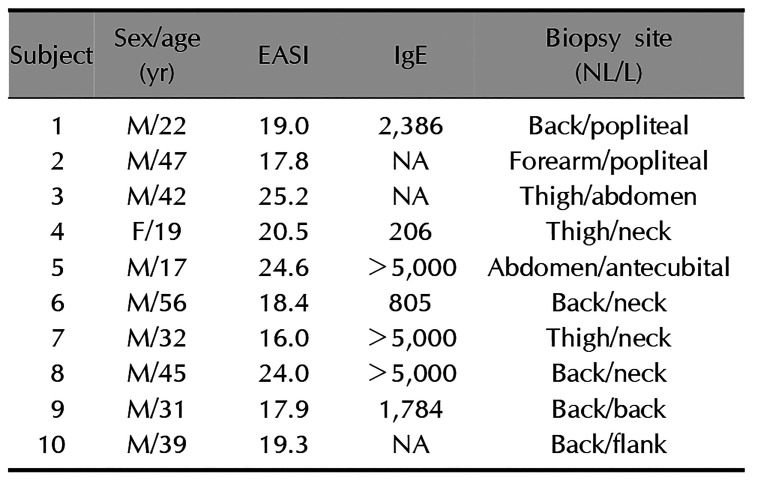
Fig. 2
Quantitative methylation analysis of the CpG 4 and CpG 2 dinucleotide units in patients with atopic dermatitis (AD) (n=10). (A) Bisulfite pyrosequencing analysis of the CpG 4 (−702 bp) and CpG 2 (−410 bp) sites in the FLG promoter region. (B) Significant association between non-lesional and lesional tissues was found in the methylation frequencies of the CpG 2 unit. The methylation frequencies of the CpG 4 and CpG 2 units were determined using triplicate experiments of bisulphite pyrosequencing. *p<0.05.
![]()
Table 1
Clinical characteristics of patients with atopic dermatitis
![]()
DISCUSSION
Epigenetic alterations in AD, including histone modifications and miRNAs, have been discovered in a variety of genes except FLG2627. The 5′-end region of FLG is devoid of any defined CGI (length ≥200 bp, Guanine-Cytosine content ≥50%, and observed-to-expected CpG ratio [ObsCpG/ExpCpG] ≥0.6) (Supplementary Fig. 2)28. The potential involvement of variable methylation in the non-CGI promoter of FLG has been mostly overlooked. In contrast to genes with CGIs at their TSSs, substantial fluctuations occur in the promoter methylation levels of genes that are CpG-poor at the TSS22. Because CpG-poor regulatory regions tend to acquire a low methylation state when occupied by transcription factors2930, DNA methylation at non-CGI promoters have high information content about the ongoing activity of transcription factors21. Several studies have shown that no significant promoter methylation is associated with FLG expression in AD cases2431 and in buccal cells32. Herein, we focused on the CpG sites that were not examined in previous epigenetic analyses, because the methylation array-based studies on FLG in AD subjects could not have included all the differentially methylated CpG loci in the non-CGI promoter of FLG. Noticeably, non-CGI promoters, which are generally hypermethylated, remain transcriptionally active regardless of their methylation state3334, but our findings demonstrate that DNA methylation of only four CpG sites directly suppress the non-CGI promoter of FLG. Moreover, significant differences were observed in the methylation frequency of the single CpG site between non-lesional and lesional epidermis obtained from Korean patients with severe AD, suggesting that the possible epigenetic alterations in vivo might depend on the methylation status at this site-specific CpG unit within the FLG promoter.
It has been previously found that the levels of filaggrin breakdown products, such as trans-urocanic acid and pyrrolidone-5-carboxylic acid, are significantly reduced in patients with AD without FLG-null mutations1735. Our results support the insight that epigenetic alterations may account for discordant phenotypes of patients with a skin disease who are wild-type on the prevalent mutations of the disease36. Because a given cytosine can either be completely methylated or unmethylated, ‘variable methylation’ is the outcome of averaging these binary states22. Hence, what is measured for a given epidermal sample at a given CpG site, i.e., the CpG 2 unit within the FLG promoter, is the percentage of epidermal cells that are methylated. Considering the significant differences in the methylation frequencies among the dermatologist-diagnosed AD samples, this key CpG 2 unit in the FLG promoter can serve as an AD-associated DNA methylation biomarker. Indeed, a single CpG site showing differential methylation between different disease states is known as a methylation variable position (MVP), which can be considered as the epigenetic equivalent of a single nucleotide polymorphism3738. This might suggest that the methylation frequency at any given MVP may serve as a novel epigenetic signature for physiological and pathologic status3940. Thus, our observations also suggest that the CpG 2 unit within the FLG promoter might serve as an MVP candidate of filaggrindeficient skin phenotype.
In conclusion, we provided evidence for one of the mechanisms via which significant contributors to filaggrin deficiency work in the lesional skin of patients with AD. To extrapolate the plausible correlation between epigenotype of FLG and genetic background of affected individuals and controls, future studies should involve a larger AD cohort where the genotyping for the FLG-null mutations is performed beforehand. Furthermore, our finding may have more promising clinical relevance in conjunction with FLG-genotyping studies, which include diseases of various severity.
 XML Download
XML Download