

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Brugada syndrome (BrS) is characterized by ST-segment elevation in the right precordial leads and associated with life-threatening arrhythmia, mainly ventricular fibrillation (VF) that leads to sudden cardiac death (SCD).1) Since BrS was described in 1992,1) a tremendous number of BrS cases have been reported its incidence, up to 1–5/10,000 worldwide.2)3) To date, the standard therapy for the prevention of SCD in BrS is the use of an implantable cardioverter-defibrillator (ICD) especially in patients who have experienced a prior cardiac arrest or syncopal events secondary to VF.4) However, ICDs do not prevent the occurrence of VF but react to defibrillate the VF episode, thereby preventing sudden death. Often patients with recurrent VF have to be maintained on antiarrhythmic drugs that have modest effectiveness and adverse effects. Many patients have to endure recurrent ventricular tachycardia (VT)/VF episodes with associated presyncope/syncope and ICD shocks that may be painful and distressing and may cause progressive heart failure and post-traumatic stress disorder. Quinidine, an antiarrhythmic drug with blocking activity in the Ito and IKr currents, has been effective in preventing VF recurrence.5) However, quinidine has undesirable side effects (thrombocytopenia, intolerable diarrhea, esophagitis, allergic reaction, aggravation of sinus node dysfunction, and the potential for QT prolongation and torsade de pointes) and its availability is limited especially in regions where BrS is endemic.6)
An alternative adjuvant therapy for BrS with recurrent VF was first described by Haïssaguerre et al.7) Eight years later, there was a paradigm shift in treatment of BrS when Nademanee et al.8) reported high efficacy of epicardial ablation to reduce VF burden. In this review, we will review the recent updates in catheter ablation for BrS.
Go to : 
ENDOCARDIAL APPROACH FOR BRUGADA SYNDROME
In BrS, VF-triggering premature ventricular complexes (PVCs) are almost identical,9) site specific,10) and predominantly originate from the right ventricular outflow tract (RVOT), which is the region hypothesized to provide slow discontinuous conduction and maximal refractoriness.8)11)12) Kakishita et al.9) demonstrated that many spontaneous episodes of VF in patients with BrS were preceded by frequent PVCs. They also found that preceding and initiating PVCs of a single and different VF episodes in the same patient showed the same pattern of QRS morphology and moreover, the QRS patterns of PVCs in 2 different patients were very similar to each other.9)
Characteristics of ventricular fibrillation-triggering premature ventricular complexes
Haïssaguerre et al.7) first reported the electrophysiological properties and effects of catheter ablation in 3 symptomatic patients with BrS, with 1 patient exhibiting a familial SCN5A deletion mutation (2850delT). Monofocal PVCs originating from the RVOT were observed in all patients, with monofocal PVCs with left bundle-branch block (LBBB) and superior axis in 1 patient. RVOT triggers were eliminated by radiofrequency catheter ablation (RFCA) at the earliest site (25 and 40 ms before QRS onset), and VF inducibility was modified after ablation in 2 patients. In the third patient, RF energy application could ablate the PVCs originating from the anterior RV Purkinje network, thus rendered the VF non-inducible. Subsequently, other anecdotal cases were published on successful ablation of triggering ectopy within the RVOT sites in patients with this condition.13)14)
Recently we reported the largest cohort of endocardial VF ablation in BrS with the longest follow-up period.15) In that cohort of 21 VF ablation cases, VF-triggering ectopic beat, documented in 12-lead electrocardiogram (ECG), exhibited LBBB configuration with inferior axis in 18 patients (Figure 1A) and superior axis in 3 patients (Figure 1B). The precordial QRS transition was seen in lead V3 in 4 patients, in V4 in 8 patients, and in V5 in 7 patients. The mean coupling interval of the clinical PVC was 415±57 ms.
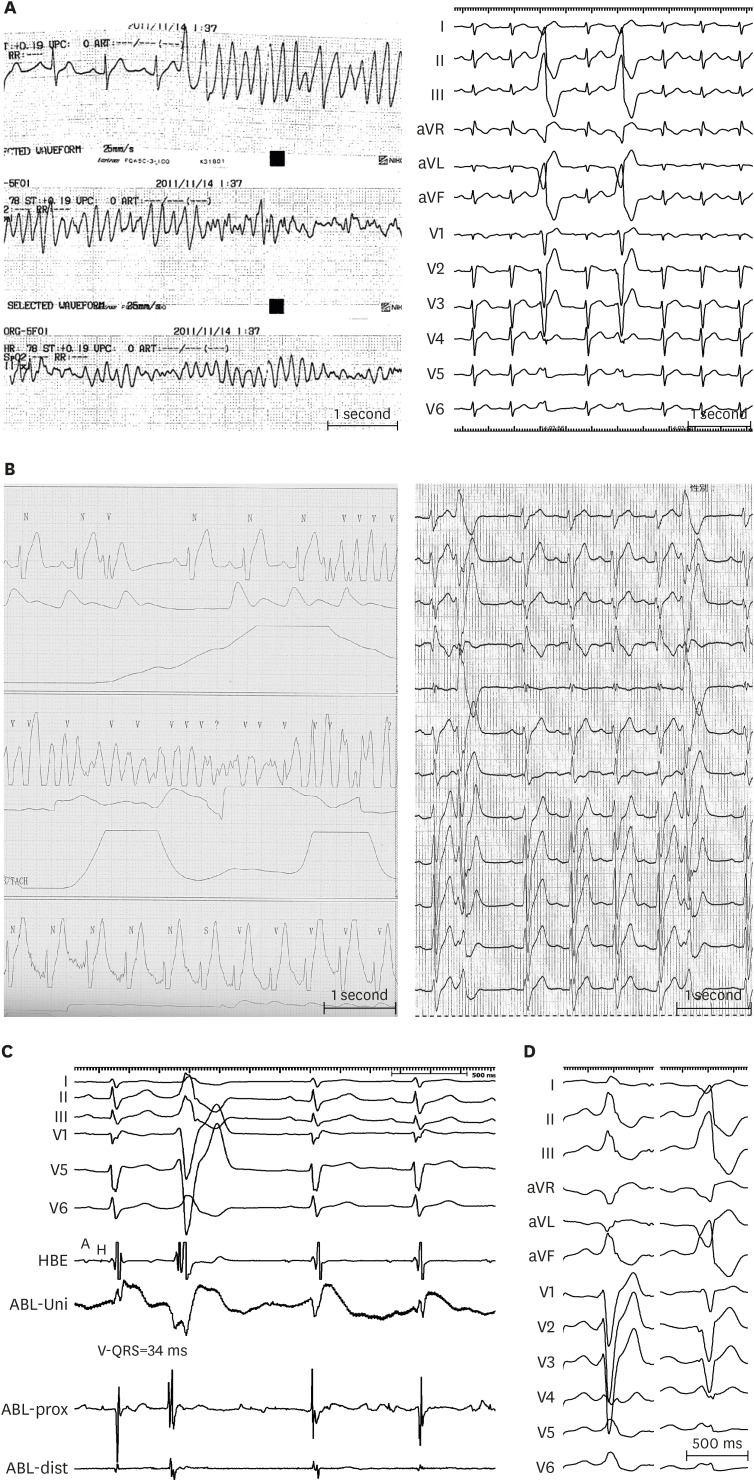 | Figure 1(A) Serial ECG strips of a BrS patient presented to the emergency room after resuscitation from VF. Frequent monofocal PVCs reproducibly degenerated into VF, leading to electrical storm. PVCs exhibited LBBB and inferior axis morphology. (B) Telemetry of a BrS patient presented with recurrent VF episodes. 12-lead ECG taken immediately after implantable cardioverter-defibrillator discharge showed PVCs with LBBB and superior axis configuration. (C) Intracardiac recordings at a successful ablation site of a VF-triggering PVC. The local ventricular activation recorded by the distal electrode pair of the ABL preceded the QRS complex onset by 34 ms with QS configuration of the unipolar electrogram. (D) VF-triggering PVC immediately after electrical storm (right) of a young male who underwent emergency VF-trigger ablation. Culprit PVC exhibits LBBB pattern and inferior axis. After culprit PVC elimination, VF became non-inducible after electrophysiology study with triple RV extra-stimuli. Pilsicainide infusion could induce a PVC of LBBB pattern, inferior axis indicating RV outflow tract origin (right); however the induced PVC morphology was different from VF-triggering PVC and was not ablated. The patient has been VF-free for 8-year follow-up.Modified from Talib AK, et al. Efficacy of endocardial ablation of drug-resistant ventricular fibrillation in Brugada syndrome: long-term outcome. Circ Arrhythm Electrophysiol 2018;11:e006675.15)
A = atrial potential; ABL = ablation catheter; aVF = augmented vector foot; aVL = augmented vector left; aVR = augmented vector right; BrS = Brugada syndrome; dist = distal bipole; ECG = electrocardiogram; H = his potential; HBE = his bundle electrogram; LBBB = left bundle-branch block; prox = proximal; PVC = premature ventricular complex; RV = right ventricular; Uni = unipolar electrogram; VF = ventricular fibrillation.
|
Ventricular fibrillation-trigger ablation in Brugada syndrome
Like any trigger ablation, ablation is performed by targeting a consistent triggering PVC inducing VT/VF, localized by mapping the earliest electrogram (EGM) relative to the onset of the ectopic QRS complex before the VF during a provocation test or by reviewing the morphology of the PVC in the rhythm strip of a spontaneous VF episode. Many case series showed that VF-triggering PVCs were spontaneously observed and successfully ablated in the RVOT in 4 cases and at the right Purkinje arborization in 1 case.7)11)12) Similarly, we found VF-triggering PVCs originated from the RVOT in 85% of the cases and from the RV itself in the rest of the cases; namely RV free wall (RVF) in 2 cases (among them only 1 case had Purkinje origin) and RV inflow tract in 1 case (Figure 1C).15)
A major limitation to trigger ablation approach is the rare occurrence of PVCs in BrS patients.16) Therefore, all efforts should be done to identify culprit PVCs. To maximize the chance of finding VF trigger, we should attempt to obtain a 12-lead ECG during the arrhythmia episodes. Noninvasive body surface mapping, such as electrocardiographic imaging, can be used from pre-ablation and during ablation to localize PVC origin. Perfect ECG documentation can be done by obtaining 12-lead ECG during or within few days of VF or electric storm because VF-triggers appear in a narrow-time window; therefore, it is important to ablate the “culprit” PVCs that initiate VF before the index PVCs subside.14)17) Sodium channel blockers such as pilsicainide is useful in inducing VF-triggering PVCs. A caveat to this approach is the induction of non-culprit PVC; therefore, it is extremely important to make sure that pilsicainide-induced PVC is the same as the culprit one.
Figure 1D shows VF-triggering PVC frequently observed immediately after electrical storm of a young male who underwent emergency VF-trigger ablation. After culprit PVC elimination, VF became non-inducible after electrophysiology study (EPS) with triple RV extra-stimuli. After ablation, pilsicainide infusion could induce a PVC of RVOT origin; however, the induced PVC was different from VF-triggering PVC and was not ablated. That patient has been event free for 8 year-follow-up.
Endocardial substrate ablation in Brugada syndrome
There is no data on endocardial arrhythmogenic substrate of BrS. Shah et al.18) first reported endocardial substrate modification in BrS. Due to the absence of clinical ectopies during EPS, endocardial ablation was performed focally on the septal and the anterolateral RVOT under the guidance of pace mapping. Initial ablation of these foci triggered flurry of non-sustained episodes of VF. Later, ectopies disappeared and ablation was reinforced in a wider area surrounding both foci (total ablation time—56 minutes). This led to the disappearance of Brugada pattern in leads V1–V2, and the ECG normalized. The ECG remained normal during the subsequent hospital stay and at every follow-up visit thereafter. The patient has not experienced any arrhythmia and the ECG has no evidence of Brugada pattern 6.5 years after ablation.18) Using noncontact mapping, Sunsaneewitayakul et al.19) suggested that the late depolarization zone on RVOT may serve as potential VF substrate in BrS patients who have frequent VF episodes and VF storms. RFCA on late depolarization zone modified ECG, suppressed VF storm, and reduced VF recurrence in BrS patients with VF storms. Hayashi et al.20) could record fractionated and delayed potentials by contact bipolar mapping of the endocardium in a BrS patient. Most of those potentials were observed within low-voltage areas in various RVOT regions.
With careful endocardial mapping, we also found that endocardial fractionated EGMs were present in the most malignant Brugada phenotype i.e., in patients with extremely frequent VF episodes who had evidence of subtle conduction delay represented by QRS notching in lead V1. Although ablating RVOT-fractionated EGMs normalized the ST-segment elevation in 3 patients (Figure 2), VF recurrence was still observed. We found that presence of QRS notch in lead V1 was invariably associated with VF recurrence despite accurate documentation of VF-triggering PVCs and extensive endocardial substrate modification, hence epicardial ablation is ultimately needed.15)
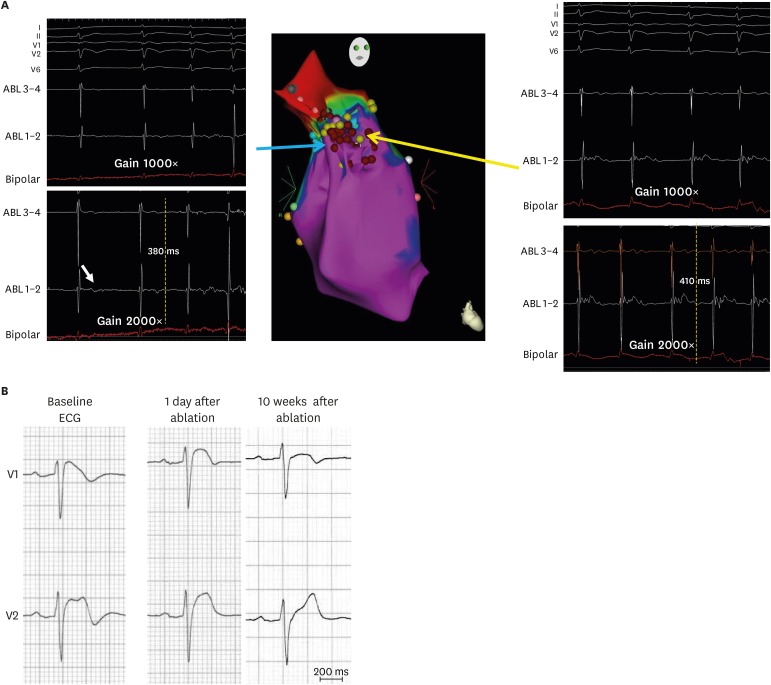 | Figure 2(A) Right anterior oblique view of 3-dimensional electroanatomical mapping of endocardial aspect of the RVOT where delayed low voltage fractionated EGMs were found at the RVOT lateral wall (red tags represent ablation points). Note these low voltage EGMs could be revealed using larger than the standard gain (2,000×) (B) Right precordial ECGs of a Brugada syndrome patient showing normalization of Brugada-type ST-segment elevation 10 weeks after endocardial ablation.Modified from Talib AK, et al. Efficacy of endocardial ablation of drug-resistant ventricular fibrillation in Brugada syndrome: long-term outcome. Circ Arrhythm Electrophysiol 2018;11:e006675.15)
ABL = ablation catheter; ECG = electrocardiogram; EGM = electrogram; RVOT = right ventricular outflow tract.
|
Go to :
EPICARDIAL APPROACH
An animal model of BrS showed that the RVOT was the main substrate site, especially at the epicardium.21) A clear clinical evidence on the importance of epicardial substrate came from a study conducted by Nademanee et al.8) who reported 9 BrS patients with frequent ICD discharges. All 9 patients had abnormal EGMs characterized by low voltage, and fractionated late potentials clustering at the anterior aspect of the RVOT epicardium.8) Ablation at these sites rendered VT/VF non inducible and normalization of the Brugada ECG pattern in the majority of patients with no recurrent VT/VF in all patients off medication except 1 patient on amiodarone.8) Moreover, an extended work by Nademanee et al.11) showed a significant increase in collagen content in all VF victims diagnosed with BrS over and above the normal collagen content seen in age-and sex-matched controls. In the same study, biopsies taken from 6 patients with BrS during open heart ablation from RVOT epicardial sites, which harbored abnormal fragmented and delayed conduction, confirmed similar findings of fibrosis. After open heart ablation at these fibrosis sites, ECG pattern was normalized, and patients no longer had recurrent VF.
One might argue that these subtle changes and abnormal fractionated EGMs may reflect an underlying RV cardiomyopathy such as arrhythmogenic right ventricular cardiomyopathy; however, all in vivo cases had normal cardiac magnetic resonance imaging and computed tomography as well as a normal appearing heart on direct visualization during thoracotomy.
Figure 3 shows an ECG of 27-year-old male with BrS and recurrent syncope. Genetic analysis revealed SCN5A mutation and pilsicainide infusion induced VF initiated by PVC. Earliest activation of the trigger PVC was observed at the endocardial surface of RVF where late potential was observed and presystolic Purkinje potential preceding the QRS onset by 35 ms. However, PVC could not be eliminated by endocardial RFCA, and the patient had still VF episodes despite maximally tolerated antiarrhythmic therapy. Endocardial electroanatomical mapping, repeated 8 years after the initial endocardial session, revealed normal RV/RVOT voltage and confirmed elimination of previously ablated endocardial EGMs. Finally, intraoperative epicardial mapping revealed relatively low voltage area with fractionated EGMs at the epicardial surface of previously ablated endocardial EGMs. Interestingly, coved-type ST-segment elevation was observed in the unipolar recordings of the ablation catheter at the fractionated EGM areas where epicardial biopsy and histology shows epicardial fibrosis with focal finger-like projections of collagen into myocardium while fractionated EGM-free areas revealed neither abnormal potential nor myocardial fibrosis. Cryoablation at the abnormal EGM sites prevented VF recurrence during further 7-year follow-up, with normalization of ST elevation in precordial leads.
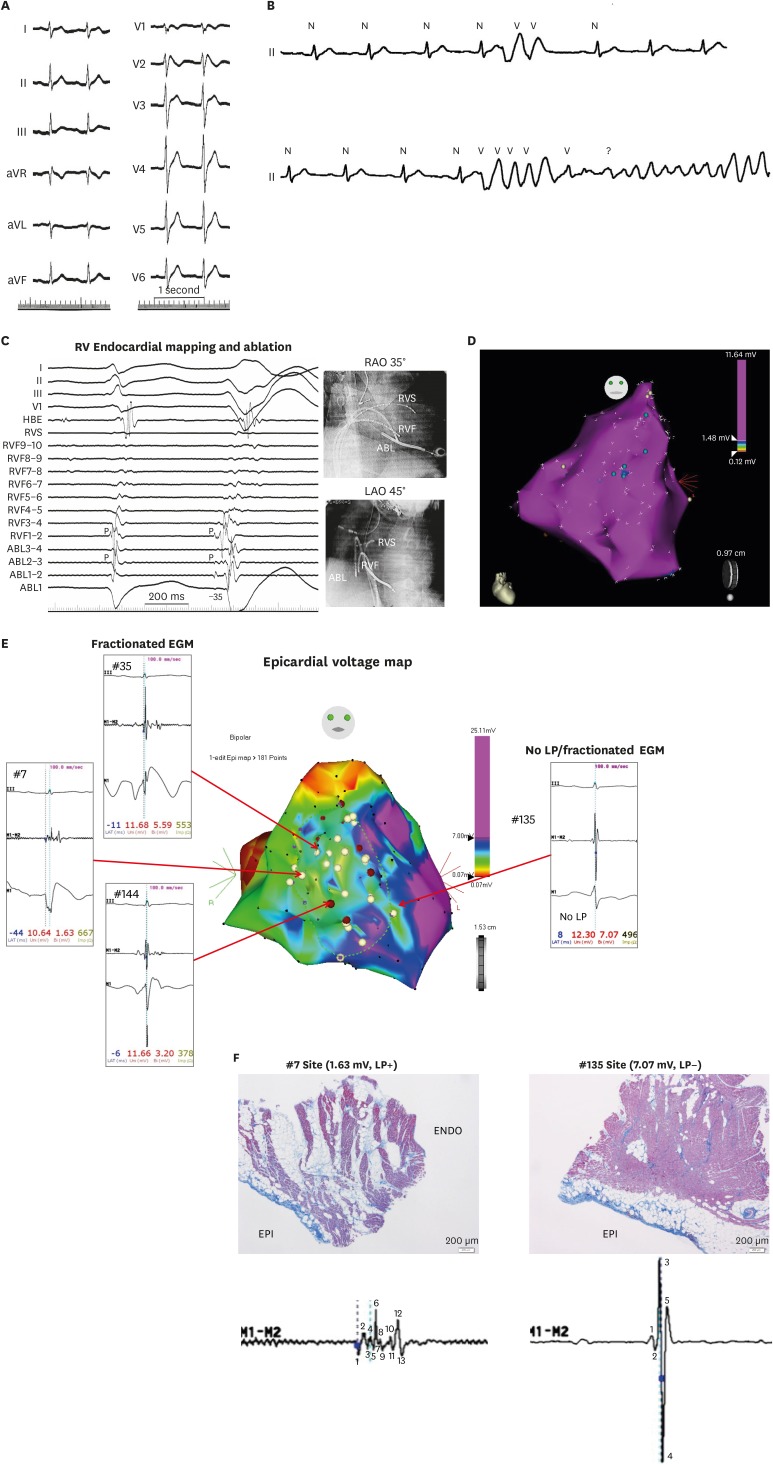 | Figure 3(A) A 12–lead electrocardiogram of a 27-year-old male with Brugada syndrome and SCN5A mutation Note QRS notching in lead V1. (B) After Pilsicainide infusion, Frequent PVCs were observed which degenerated into VF. (C) Mapping showed the earliest activation of the VF-triggering PVCs were observed at the RVF where presystolic Purkinje potential preceding the QRS onset of the PVC by 35 ms. Note that Purkinje potential is observed locally during sinus rhythm. (D) Endocardial electroanatomical mapping, repeated 8 years after the initial endocardial session, revealed normal RV/RV outflow tract voltage and confirmed elimination of previously ablated endocardial EGMs. Blue tags indicate remnant previously ablated LP. (E) Intraoperative epicardial mapping revealed relatively low voltage area with fractionated EGMs at the epicardial surface of previously ablated endocardial EGMs. Interestingly, coved-type ST-segment elevation was observed in the unipolar recordings of the ablation catheter at the fractionated EGM areas. (F) Epicardial biopsy and histology shows epicardial fibrosis with focal finger-like projections of collagen into myocardium while EGM-free areas revealed neither abnormal potential nor myocardial fibrosis.Modified from Talib AK, et al. Efficacy of endocardial ablation of drug-resistant ventricular fibrillation in Brugada syndrome: long-term outcome. Circ Arrhythm Electrophysiol 2018;11:e006675.15)
ABL = ablation catheter; aVF = augmented vector foot; aVL = augmented vector left; aVR = augmented vector right; EGM = electrogram; HBE = his bundle electrogram; LAO = left anterior oblique; LP = late potentials; PVC = premature ventricular complex; RAO = right anterior oblique; RVF = RV free wall; RV = right ventricular; RVS = RV septum; VF = ventricular fibrillation.
|
Epicardial substrate characteristics
Definition of abnormal epicardial substrate is still arbitrary. The most common definitions for the Brugada substrate were low voltage areas <1.5 mV, late potentials, and fragmented potentials. Nademanee et al.8) defined epicardial substrate as low voltage areas <1.0 mV; split EGMs or fractionated EGMs; wide duration >80 ms or late potentials extending beyond the end of the QRS. These sites were exclusively localized over the anterior aspect of the RVOT epicardium. Brugada et al.22) defined abnormal substrate as areas with amplitude less than 1.5mV or associated wide duration (>80 ms), multiple (>3), or delayed components extending beyond the end of the QRS complex. Epicardial areas were variable in extension and distribution mainly located in the RVF and RVOT.22) Pappone et al.23) defined abnormal substrate as low-frequency (up to 100 Hz) prolonged duration (>200 ms) bipolar signals with delayed activity extending beyond the end of the QRS complex.
Methods to enhance epicardial functional substrate
Clinically, sodium channel blockage is an essential diagnostic test to unmask BrS. Such provocation test has important clinical implications in the EP lab to enhance epicardial functional substrate. Using high density mapping, Sacher et al.24) found a clear correlation between epicardial late potential and fragmented EGM splitting with appearance of Brugada type ECG during ajmaline infusion in a case of BrS with aborted SCD.
Recently, Brugada et al.22) proposed flecainide infusion (2 mg/kg per 10 minutes) as a novel method to accurately determine the location and size of arrhythmic electrical substrate (AES) in a series of 14 patients of BrS. Pappone et al.23) found that AES is commonly located in the right ventricle epicardium and ajmaline exposes its extent and distribution, which is correlated with the degree of coved ST-elevation in a series of 135 patients. Ajmaline infusion revealed a significant increase in BrS substrate size, even though most patients with the worst clinical presentation did not have a baseline spontaneous type 1 BrS ECG pattern, a family history of SCD at age<45 years, or a positive test for SCN5A.23) More recently, Chung et al.25) found that epicardial warm water instillation in patients with BrS could result in the augmentation or the presence of electrocardiographic phenotype, enhance epicardial functional substrates owing to the heterogeneous variation in conduction velocity, and facilitate the ventricular arrhythmogenesis.
Figure 4 shows electro-anatomical map of epicardial substrate of a 22-year-old male with BrS. Baseline voltage map showed low voltage areas at the anterior and lateral aspects and RV and RVOT (left). After pilsicainide infusion, the low voltage areas extended to involve almost the entire anterior and lateral aspects of RV and RVOT (right). Demonstration of epicardial activation using ripple map also showed more delayed activation after using Pilsicainide infusion compared to the baseline epicardial activation (Supplementary Video 1).
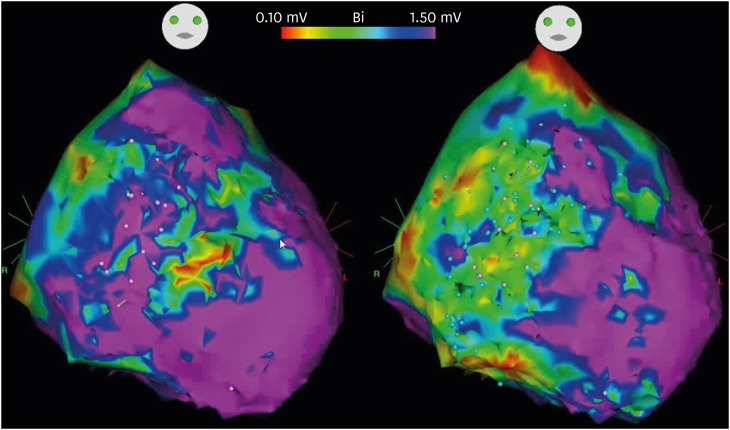 | Figure 4Electro-anatomical map of epicardial substrate of a 22-year-old male with Brugada syndrome. Baseline voltage map showed low voltage areas at the anterior and lateral aspects and RV and RVOT (left). After Pilsicainide infusion, the low voltage areas extended to involve almost the entire anterior and lateral aspects of RV and RVOT (right).RV = right ventricular; RVOT = right ventricular outflow tract.
|
Epicardial ablation endpoint
VT/VF non-inducibility or normalization of the Brugada ECG pattern during the ablation procedure was the initial epicardial ablation endpoint.8) However, with more understanding of epicardial substrate and longer follow up data, it became clear that the ideal end point is to eliminate all substrate areas with low voltage fractionated signals/late potentials that can be detected by sodium channel blockade. An example is Supplementary Video 2 where baseline voltage map showed low voltage areas at the anterior and lateral aspects and RV and RVOT (left). After ablation, the low voltage areas extended to involve almost the entire anterior and lateral aspects of RV and RVOT (right) with ripple map showing only remnant late epicardial activation after ablation. Nowadays ablation is performed with irrigation catheter and preferably with contact force-based catheters that enable good epicardial contact and direct the catheter vector toward the epicardium with careful monitoring of the local EGM which can be delayed, diminished or eliminated. However, achieving "complete" substrate elimination can still be challenging.
A caveat to this approach is presence of early repolarization syndrome combined with Brugada ECG. Such patients may harbor additional VF risk even if RV/RVOT substrate elimination/modification is achieved with guide of sodium channel blockers. An example is exhibited in Figure 5 for a young male who presented with electrical storm. Baseline ECG showed J-wave in leads V3–V6 and after very low dose pilsicainide infusion, coved type ECG pattern was observed resulting in global type early repolarization (left). Despite extensive epicardial ablation at the RV/RVOT with sodium channel blockade guide, Brugada EG pattern could be eliminated even after higher dose pilsicainide; however, lateral chest lead J-wave could not be eliminated. Such patients may remain at high risk of developing VF after ablation.
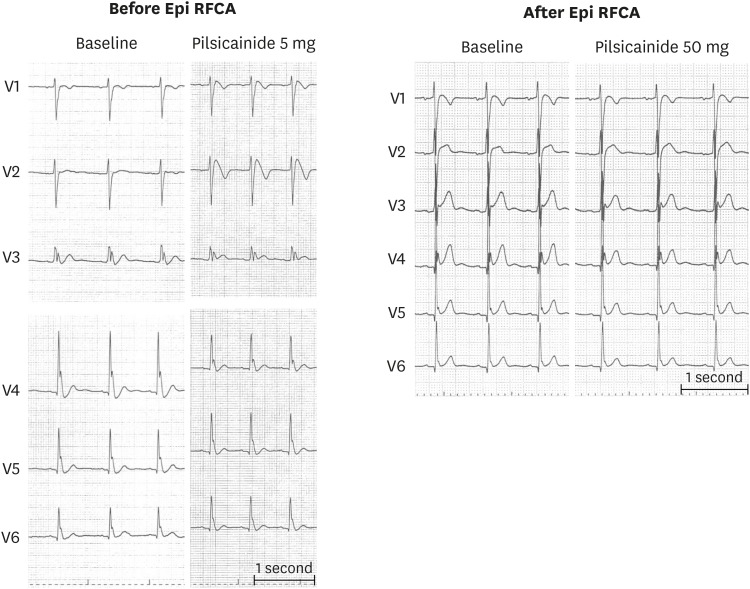 | Figure 5A 12-lead ECG of a young male who presented with electrical storm. Baseline ECG showed J-wave in leads V3–V6 and after very low dose (5 mg) Pilsicainide infusion, coved type ECG pattern was observed resulting in global type early repolarization (left). Despite extensive epicardial ablation at the RV/RV outflow tract with sodium channel blockade guide, Brugada ECG pattern could be eliminated even after higher dose Pilsicainide however, lateral chest lead J-wave could not be eliminated (right). Such patients may remain at high-risk of developing VF after ablation.ECG = electrocardiogram; RFCA = radiofrequency catheter ablation; RV = right ventricular; VF = ventricular fibrillation.
|
With the evidence we have so far, we think that combined approach of VF-trigger elimination if trigger ECG is well documented and non-inducibility, ECG normalization and most importantly sodium channel blockade-guided substrate elimination should all be confirmed if possible.
Go to :

 XML Download
XML Download