

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Calcium (Ca) and phosphate (Pi) are important elements in sustaining life. Ca is mainly extracellular (99%) and concentration changes can be monitored with relative ease, in striking contrast with Pi that only 1% of its total presence is in the extracellular fluid.[1] Ca concentration in plasma and extracellular fluid is remarkably constant. Through the day it is estimated that serum Ca fluctuates only by 0.25 mM in normal individuals. On the other hand, plasma Pi concentration varies considerably through the day and is significantly affected by diet.
Their concentration is regulated mainly by 3 hormones, the parathyroid hormone (PTH), 1,25-dihydroxy-vitamin D (1,25[OH]2D) and, with the presence of klotho, fibroblast growth factor-23 (FGF-23). They co-ordinate the release of these 2 elements from the skeleton where most of these elements are, their absorption from the food in the intestine and their re-absorption/excretion in the kidney (Fig. 1).[2]
 | Fig. 1Simplified schematic presentation of the interactions of parathyroid hormone (PTH), 1,25-dihydroxy-vitamin D (1,25[OH]2D), and fibroblast growth factor-23 (FGF-23) that regulate the calcium (Ca) and phosphate (Pi) homeostasis. The parathyroid glands operate as the command center. PTH induces 1,25(OH)2D and FGF-23 production, increases Ca re-absorption and Pi excretion in the kidney and stimulates their release from bone; 1,25(OH)2D increases Ca and Pi absorption in the intestine, and FGF-23 levels, but have inhibitory effect on PTH synthesis; FGF-23, in the presence of the obligate co-receptor α-klotho, inhibits Pi reabsorption in the kidney, increases production and catabolism of 1,25(OH)2D and production and secretion of PTH. IGF-1, insulin-like growth factor 1.
|
The relationship between Ca (via the Ca sensing receptor [CasR]), Pi, PTH, 1,25(OH)2D and FGF-23 have been previously well described.[3] Briefly, there is an inverse sigmoidal relationship between [Ca2+] and PTH secretion rate.[4] In the skeleton, PTH increases the rate of bone remodelling and its action becomes anabolic when its excretion is pulsatile, and catabolic when the bone is exposed to continuously high PTH levels. In the kidney, PTH increases Ca reabsorption, Pi excretion and 1,25(OH)2D and FGF-23 production.[5] As part of the feedback loop, 1,25(OH)2D suppresses PTH production. It also increases Ca and Pi absorption in the intestine.[6] FGF-23, a phosphaturic hormone, decreases circulating Pi, 1,25(OH)2D [78] and PTH mRNA.
Any factors disturbing this delicate homeostatic system could cause severe clinical complications. In renal impairment, the effects are even more obvious because of the unique role of the kidney which is the only place where the body has the option to act selectively and keep in the system only the amount of Ca and Pi needed.
The Ca and Pi dysregulation that follows the loss of nephrons, with Pi retention being accepted as the initiating event, [9] leads to a complication which is nearly universal in moderate and advanced stages of renal impairment, known as mineral and bone disorders of chronic kidney disease (CKD-MBD). This term was introduced in 2005 to describe the broader clinical syndrome encompassing mineral, bone, and calcific cardiovascular abnormalities that develop as a complication of CKD. Renal osteodystrophy is a component of CKD-MBD. It defines alterations in bone histology associated with CKD, based upon bone turnover, mineralization and volume, and provides a histomorphometric classification for the spectrum of heterogeneous bone diseases associated with loss of renal function.[10]
The spectrum of renal osteodystrophy consists of the predominant hyperparathyroid-mediated high-turnover bone disease (osteitis fibrosa), and secondary hyperparathyroidism (HPT), is a routine finding by stage 3A CKD; osteomalacia (defined as a mineralization lag time greater than 100 days); low turnover osteomalacia (defective mineralization in association with low osteoclast and osteoblast activities); mixed uremic osteodystrophy (hyperparathyroid bone disease with a superimposed mineralization defect), and the iatrogenic and idiopathic adynamic bone (diminished bone formation and resorption), but not osteoporosis.[11]
Unlike osteoporosis, CKD-MBD is primarily a disturbance of mineral homeostasis that leads to metabolic bone diseases. Therefore, the pathophysiology of renal osteodystrophy (and the role of PTH) is completely different to that of osteoporosis where PTH levels remain normal throughout (as is the case with serum Ca and Pi concentrations as well). Furthermore, an intervention in patients with CKD that could modulate the hormones regulating Ca and Pi concentrations would help restore mineral homeostasis, in contrast to an intervention in patients with osteoporosis that would help restore the damaged bone architecture.
It is also useful to recognize the separation of the homeostatic from the remodeling system. It is widely believed that the plasma [Ca2+] is regulated by the rate of bone resorption, a belief that has been comprehensively refuted.[12] In a healthy individual, plasma Ca reflects the level of equilibration at quiescent bone surfaces between systemic and bone extracellular fluid set by PTH. Bone remodeling is not directly concerned with the regulation of plasma Ca and makes a less direct contribution to Ca homeostasis.[12] Primarily, it is the Ca transport in the distal nephron and the exchange of Ca between plasma and a rapidly exchangeable pool in bone that determine the min-to-min changes in PTH release into the circulation (Fig. 2).[1314]
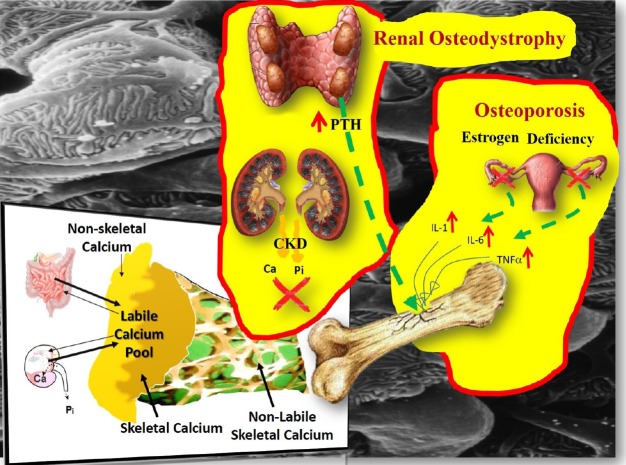 | Fig. 2Instant min-to-min (labile calcium [Ca] pool) and bone remodeling contribution to Ca and phosphate (Pi) homeostasis. The labile pool of Ca2+ can buffer an acute Ca load as well as to maintain a stable Ca concentration during acute Ca deprivation. The magnitude of the rapid Ca exchange was estimated to be many fold higher than the daily flux from remodeling based bone turnover.[70] In postmenopausal osteoporosis, provided that the renal function is normal, the minerals released from the skeleton following the loss of bone mass, do not stretch the buffering capacity of the labile pool beyond its limits, because the kidney responds to the challenge and the blood levels of Ca, Pi, parathyroid hormone (PTH), fibroblast growth factor-23 and 1,25-dihydroxy-vitamin D remain unaffected. In bone disorders of chronic kidney disease (CKD), because of the loss of renal mass and function, it is the labile pool that is affected first, before the bone and the parathyroids (secondary hyperparathyroidism) get involved.
|
The clinical management of CKD-MBD is problematic because current therapies do not fully correct the abnormalities leading to its development and progression.[13] It is remarkable that in the management of patients with CKD, and especially those not yet on dialysis, there is no major breakthrough since the introduction of calcitriol (1,25[OH]2D3) in the late ′70s. An unexplored so far option is the intermittent administration of PTH that could offer a novel approach in the management of CKD-MBD in the clinic. Contrary to the current practice where pharmaceutical intervention (typically Pi binders) is considered at advanced, pre-dialysis stage, administration of PTH initiated as soon as the FGF-23 concentration raises above the normal range limits, would target the very first step of dysregulation of Ca and Pi homeostasis, well before this imbalance becomes a bone complication.
The aim of this article is to highlight the most important aspects of the pathophysiology of CKD-MBD and its current management and propose the use of intermittent PTH administration in the prevention and treatment of CKD-MBD.
Go to : 
MINERAL AND BONE DISORDERS OF CHRONIC KIDNEY DISEASE
There is a high prevalence of CKD in the general population. In the USA it is estimated that the lifetime risk of having a glomerular filtration rate (GFR) of <60 mL/min per 1.73 m2 is 59% [15] or approximately 7.2% of the adult population has stage 3 to 5 CKD.[16] In England, data from the 2009 to 2010 Health Survey and the 2011 Census projected that in 2011, 2.6 million or 6.1% of the population aged 16 or older had stage 3 to 5 CKD.[17] The majority of these patients have already developed MBD. CKD-MBD could start relatively early when the GFR is in the region of 60 to 90 mL/min as shown by abnormalities found on bone histology or elevated PTH levels.[181920] Even in patients with normal PTH concentrations, bone histology could be abnormal.[18]
1. The natural history of renal osteodystrophy
Our attempts to understand the genesis of CKD-MBD and dissect the pathophysiology of renal osteodystrophy, mainly the development of secondary HPT, revolve around the almost half-century old “trade-off hypothesis” which implicates Pi retention as the initiating event in the disruption of homeostasis.[9] More specifically, Pi retention has been accepted as the primary driving force initiating CKD-MBD. In the early stages of renal impairment, clinically and experimentally, normal serum Pi concentrations is a common finding. Due to the compensatory increase in FGF-23, hyperphosphatemia begins to be noticeable in advanced renal insufficiency. Recent studies showed that in the early stages of renal impairment the first detectable change is a significant elevation of serum concentrations of FGF-23,[2122232425] leading to increased Pi excretion through the kidneys. Thus, elevated FGF-23 levels suggest already increased levels of Pi and therefore support a central role for Pi retention. Yet, on the same token, it does not necessarily establish the changes in serum Pi concentration as the exclusive initiating event in the dysregulation of mineral homeostasis.
The presiding “trade-off hypothesis” and the “dogma” of Pi retention as the primary pathogenetic factor in the disruption of homeostasis, still remain the leading direction of research and clinical practice, focused on the extracellular levels of Pi. The outcome has been mixed, rather disappointing, and, unfortunately, we are still far from reaching the stage where we could treat effectively the MBD of CKD.[26]
2. Current therapeutic approach
Pi binders are the mainstay of therapy and are routinely prescribed in dialysis patients. In the gastrointestinal tract, they form insoluble, non-absorbable complexes. Over the years, several preparations have been introduced. Aluminum-based binders are the most effective but their association with skeletal (osteomalacia), hematological (macrocytic anemia) and neurological adverse events (dementia) in the late ′70s restricted their use for short periods only to those with poorly controlled hyperphosphatemia after treatment with other binders has failed. The most widely used Ca-based binders could increase the risk of Ca overload and accelerate vascular calcification; the risk of adynamic bone disease could be increased as well.[27] Ca-free, aluminum-free, binders include non-absorbed polymers (Sevelamer), bivalent and trivalent cation binders (Lanthanum salts and Magnesium and Iron salts).
The effect of Pi binders on serum Pi and FGF-23 levels is relatively small.[28] Furthermore, their effects on patient-important outcomes compared to placebo are uncertain. In patients with CKD Stage 2 to 5 (G2 to G5), their effects on cardiovascular, vascular calcification, and bone outcomes compared to placebo or usual care, are also uncertain.[29] Overall, the evidence is lacking to demonstrate the efficacy of Pi binders for lowering serum Pi in patients with CKD G3a to G4 and the safety of Pi binders in this population is unproven.[30]
Historically, the first therapeutic landmark was the introduction of calcitriol in the late seventies, early eighties. It raised hopes that by increasing the levels of the active metabolite of vitamin D 1,25(OH)2D we will be able to restore the homeostatic balance of Ca and Pi.[31] Despite an initial improvement, the development of hypercalcemia and hyperphosphatemia sooner or later may force the discontinuation of treatment. New vitamin D analogues or derivatives, claiming to be less potent on Ca and Pi absorption in the intestine, have been introduced, although their effectiveness is also limited.[32] Therapy with these agents may have additional harmful effects related to increases in serum Pi and FGF-23 levels. Therefore, the 2017 Update Work Group recommended that the use of calcitriol or vitamin D analogues should be reserved only for severe and progressive secondary HPT and no longer recommends routine use of calcitriol or its analogues in CKD G3a to G5.[30]
A new class of agents, the Calcimimetics, target the CasR in the parathyroids and reduce PTH levels and parathyroid cell proliferation and thus improve the hyperparathyroid status and delay the development of tertiary HPT. They trick the CasR to think that the serum Ca is higher than it is and thereby reduce parathyroid cell PTH production. They, alone or in combination with calcitriol or any of its analogues, have become part of the routine practice in patients on dialysis (not those who are not yet on dialysis) where other therapeutic approaches have failed. However, they are far from the solution to this huge clinical challenge.[33]
The recent discovery of FGF-23 brought a lot of excitement [34] but still, the gaps left by the trade-off hypothesis have not been fully filled. And even more important, following its discovery, there are no therapeutic gains for the clinicians and the patient. Elevation in FGF-23 is to mainmaintain normal mineral and hormonal homeostasis. Therefore, monoclonal antibody treatment against FGF-23 is not the answer because we deprive the system from a powerful phosphaturic hormone and allow the development of hyperphosphatemia and therefore, it could well be that we simply kill the “messenger” without solving the problem. Indeed, neutralization of FGF-23 led to sustained reductions in secondary HPT, including decreased PTH, increased vitamin D and serum Ca concentrations, as well as normalization of bone markers such as cancellous bone volume, trabecular number, osteoblast surface, osteoid surface, and bone-formation rate. However, this was followed by an increased risk of mortality in the CKD-MBD rats treated with FGF23-Ab, most likely attributed to the dose-dependent increases in serum Pi and aortic calcification.[35]
The possibility that FGF-23 could be a “uremic toxin” has also been raised. Observational studies have suggested that elevated FGF-23 could increase significantly the risk of cardiovascular events including congestive heart failure [36] and left ventricular hypertrophy (LVH),[373839] or it could be a detrimental factor affecting survival and a predictor of mortality. It could be argued as well that FGF-23 is indirectly involved on cardiac remodeling. This could be due to its properties as sodium-conserving hormone. In addition to its action on sodium-dependent Pi cotransporters Napi2a and Napi2c, in the distal tubule FGF-23 upregulates the sodium-chloride cotransporter (NCC) and thus facilitates sodium resorption.[40] It is still debated however whether FGF-23 is simply a marker of increased concentrations of Pi. A most recent systematic review and meta-analysis of the evidence from prospective studies for associations between FGF-23 and the risk of different cardiovascular diseases concluded that the relationship between FGF-23 and cardiovascular disease risk may be non-causal.[41] Furthermore, LVH is not a prominent feature in patients with FGF-23 mediated hypophosphatemic disorders. Also, there is no evidence that inhibition of FGF-23 offers a survival advantage.[42]
3. Ca retention in the Genesis of CKD-MBD: a factor we ignored for too long
Loss of renal function results in reductions of Ca and Pi excretion. In individuals with normal renal function, the fractional urinary excretion of Ca is only 1% to 2% of the ultrafilterable Ca (ionized and complexed fractions) and the amount of Pi excreted in the urine is approximately equal to that absorbed from the intestine. The first GFR decrement in the course of progressive renal disease will raise both serum Ca and Pi levels. These relatively small elevations are less likely to promote CaHPo4 complex formation leading to drops in serum [Ca2+] which will trigger PTH secretion to increase Pi excretion through the kidneys.[43] The sustained normocalcemia (albeit at slightly higher levels but still well inside the normal range), could initially prevent any elevations of PTH. Concurrently, FGF-23 could intervene to correct the Pi levels and prevent the development of hyperphosphatemia. This new pattern of response to Ca and Pi retention establishes FGF-23 as the dominant hormone in the regulation of Pi concentrations. Furthermore, FGF-23 will have inhibitory effects on PTH secretion whilst extracellular Ca2+ remains within normal range.[44]
At some stage, because of constantly raised Pi levels, FGF-23 will reach its limits on normalizing serum Pi and this could be the point when PTH will start rising, thus establishing a new equilibrium status in the homeostatic system. The new biochemical setting will become clinically obvious with the establishment of secondary HPT, high serum Pi, low/low normal Ca, high PTH and histologically, parathyroid hyperplasia. FGF-23 levels will remain elevated because of the hyperphosphatemia; 1,25(OH)2D concentrations concentrations will remain suppressed, also due to hyperphosphatemia and high FGF-23 levels.
The inclusion of Ca retention, in addition to Pi retention, in the currently accepted trade-off hypothesis, would expand the working frame of the pathophysiology of the genesis of CKD-MBD, and improve our understanding of the sequence of events leading to the development of secondary HPT and most important, it could promote an effective approach in the clinical management of this complication.
Early administration of PTH could decrease the TmP/GFR, leading to excretion of the retained portion of Pi and keep the PTH dominant in the regulation of Ca and Pi homeostasis. Given in appropriate amounts and intervals, the levels of Pi could be controlled adequately without the need for compensatory higher FGF-23 levels. Thus, by preventing the increase of FGF-23, the decline in 1,25(OH)2D concentration could be avoided as well. At the same time, the exogenous PTH could prevent the development of hyperparathyroid bone disease and hypertrophy of the parathyroid glands, and skeletal resistance to PTH.[45] Furthermore, given intermittently, it is likely to produce anabolic response instead of the detrimental catabolic effects of the constantly raised PTH of the secondary HPT (Fig. 3). Administration of exogenous PTH can still be beneficial to bone and anabolic despite a mild elevation of endogenous PTH in patients with osteoporosis (personal communication-P. Miller).

Improvement in the Ca and Pi homeostasis, if could be achieved by Intermittent PTH administration, it could prevent or reduce the development of vascular calcification [46] which commences at the early stages of CKD-MBD,[47] and is a common finding in patients with end-stage renal disease (ESRD) and responsible for increased incidence of cardiovascular events leading to fatal outcomes.[48495051]
The timing of initiating treatment in CHD-MBD is critical. Kidney Disease Improving Global Outcomes (KDIGO) guidelines, particularly in the case of CKD patients not on dialysis, recommend initiating treatment only in the event of “progressive or persistent hyperphosphatemia,” and not for the prevention of hyperphosphatemia.[30] Indeed, currently, the primary goal of CKD-MBD therapy is the suppression of elevated levels of PTH.[52] However, there is evidence that even serum Pi levels within the normal range are associated with adverse patient outcomes and thus waiting for patients to develop overt hyperphosphatemia may not be the correct approach.[2753] In fact, there have been calls to initiate therapy in CKD G3 (GFR <60 mL/min per 1.73 m2) to prevent the progressive complications of CKD–MBD, when FGF-23 and PTH levels first begin to increase, and before hyperphosphatemia and hypocalcemia appear, albeit with Pi binders and vitamin D analogues.[5455] Furthermore, attention has been drawn to the need for potential biomarkers such as FGF-23 or the urinary fractional excretion of Pi for early diagnosis and possible treatment of disordered Pi metabolism before end-organ damage occurs.[275356]
Intervention at an early stage is the preferred option because a preventive approach could provide the maximum benefit and impact as opposed to treating an established pathology. If vigorous clinical testing confirms the anticipated beneficial effects following intermittent administration of PTH, it is sensible to consider administering PTH as soon as the FGF-23 concentration raises above the normal range limits (Fig. 4).
 | Fig. 4Intervention with phosphate binders and vitamin D analogues at advanced stages of chronic kidney disease (blue arrow) is the current clinical practice. We propose intermittent administration of parathyroid hormone at a much earlier stage (red arrow), soon after the first detection of elevated fibroblast growth factor-23 levels [Modified from “Forging forward with 10 burning questions on FGF23 in kidney disease.”, by Wolf M., 2010, J Am Soc Nephrol, 21, pp. 1427–1435. Copyright 2010 by the Williams & Wilkins. Modified with permission].
|
The determination of the dose and intervals between administrations requires carefully designed studies. Interestingly, the pharmacokinetics of teriparatide in dialysis-dependent CKD have been reported to be similar to those observed among postmenopausal women with normal kidney function, likely due to hepatic clearance of the drug.[57] As Ca and Pi concentrations remain normal at the early CKD stages, the studies should be based on baseline FGF-23 and PTH plasma concentrations with normalization of FGF-23 the primary target. Bone specific alkaline phosphatase activity should also be taken into account. Radiological evidence such as hand X-ray findings and other imaging test or biochemical markers of bone turnover could be of additional help in the overall assessment of the patients.
Overall, the amount of PTH administered may vary according to the stage of CKD of each individual patient at the time of the assessment by a clinician. Higher amounts of PTH should be required for patients at more advanced stages of CKD. Furthermore, the dosage regimen will be affected by the diet; for example, patients with higher Pi intake in their daily diet will require more frequent PTH administration.
Intermittent PTH could be administered in patients at different stages of renal impairment not yet on dialysis. Even in ESRD in patients with 2° HPT (not tertiary), intermittent administration of PTH could reduce the increased rate of Ca and Pi release from the skeleton, prevent the development of the hyperparathyroid state, and could also decrease FGF-23 production. Teriparatide or biosimilars restricted to the N-terminal PTH (1–34), the active part of PTH, are the preferred agents because longer molecules are expected to metabolize and produce fragments of unknown function. The required dose could be less than the 20 mcg and the administration could be less frequent compared to the daily use for osteoporosis. However, we should not dismiss the possibility that skeletal resistance to PTH in CKD (perhaps due to increases in sclerostin or changes in PTH-R1 expression) might reduce PTH efficacy or require higher doses.
Another option could be the PTH-related protein (PTH-rP) analogue abaloparatide approved recently by the FDA for treatment of postmenopausal women with high risk osteoporosis. It shares 76% amino acid sequence identity with human PTH-rP (1–34) and 41% with human PTH (1–34). Abaloparatide binds to PTH1R with higher selectivity for RG rather than R° confirmation of the PTH1R, resulting in transient stimulation of the receptor. This leads to greater overall anabolic effect than teriparatide. Abaloparatide undergoes non-specific proteolytic degradation and the resulting peptide fragments are eliminated by the kidneys. The recommended dosage is 80 mcg subcutaneous injection once daily.[58] Abaloparatide, compared to teriparatide, might have an advantage of being less likely to cause hypercalcemia.
Increased cortical porosity has been described in patients with osteoporosis treated with PTH. It has not, however, been shown to alter bone mechanical strength or increase the risk for hip fractures in clinical trials.[59] In patients with early secondary HPT, intermittent PTH administration could be able to bring the continuous endogenous PTH production back to its baseline. If the dose and frequency of the administered PTH in CKD-MBD cases is lower and with longer intervals compared to those for osteoporosis, then there is a good chance that it could cause less cortical porosity. In patients at more advanced CKD stages, intermittent PTH administration may not make the existing secondary HPT (parathyroids and bone) worse and, at least, it could stop its progress. However, although all these beneficial effects are desirable outcomes, we may see, instead, negative effects occurring with PTH treatment. In CKD patients with high turnover bone disease, for example, PTH treatment could lead to CKD-associated osteoporosis by increases in cortical porosity and thinning due to endocortical trabecularization.
Also, we could consider using abaloparatide which has not shown evidence of any increases in cortical porosity in rodent or primate animal models,[6061] and compared to teriparatide, abaloparatide decreased bone mineral density to a lesser extent at the 1/3 radius (primarily cortical bone). [62] Another potential advantage in using abaloparatide could be the reported increase in bone forming activity without increasing bone resorption.[63]
4. The use of PTH in CKD
PTH 1–34 (Teriparatide) has been used off-label for the treatment of adynamic bone disease in a limited number of cases with promising results. Adynamic bone disease is defined by markedly low-bone turnover resulting from a reduced number of osteoclasts and osteoblasts without osteoid accumulation. There is no consensus on its definition and diagnosis.[64] Iatrogenic interventions i.e. over-treatment of HPT, are strongly associated with its emergence. In addition, there have been suggestions that resistance to the action of PTH, high serum sclerostin levels, uremic toxins or repression of the osteocyte Wnt/β-catenin signaling pathway could cause adynamic bone disease in early stages of CKD.[64] Intermittent administration of PTH increased bone formation and bone turnover and trabecular bone mass.[65]
In experimental renal failure, intermittent administration of PTH (1–34) protects against bone demineralization and vascular calcification. In an animal model with established advanced CKD following 5/6 nephrectomy, the intermittent administration of PTH (1–34) increased bone formation and expression of renal but not bone PTH1R mRNA, while decreasing vascular calcification.[66]
A recently published study provided evidence supporting the notion that intermittent administration of PTH is safe and effective in late-stage CKD and secondary HPT. In a rat 5/6 nephrectomy model (stage 4 CKD), after 4 weeks of treatment, Ca and Pi concentrations did not change and there was a non-significant trend of lower intact PTH and FGF-23 in the teriparatide treated animals. Furthermore, teriparatide treatment showed anabolic action even under secondary HPT. It increased the bone turnover and significantly increased the degree of mineralization, micro-geometry, and bone volume, resulting in a significant improvement in mechanical properties.[67]
In the teriparatide Fracture Prevention Trial, a double-blinded placebo-control trial of 1,637 ambulatory postmenopausal women ranging in age from 42 to 86 years, 45% of patients had renal impairment (serum creatinine ≤2 mg/dL). The patients were treated with teriparatide 20 mcg/day (or 40 mcg/day) and daily Ca (1,000 mg) and vitamin D (400–1200 IU) supplementation. The incidences of treatment-emergent and renal-related adverse events were consistent across treatment groups in the normal and mild and moderate baseline renal function impairment categories. Among patients treated with teriparatide 20 mcg/day, the incidence of 4 to 6 hr post-dose serum Ca concentrations >10.6 mg/dL was significantly higher in those patients with moderate renal impairment versus those with normal renal function. Also, teriparatide therapy was associated with an increased incidence of hyperuricemia in a dose-dependent fashion. However, this increased incidence of hyperuricemia was not associated with increases in related adverse events such as gout, arthralgia, or nephrolithiasis.[68]
Similar findings were reported in real-world clinical settings. A post hoc analysis of a post marketing surveillance study of 1,847 patients with osteoporosis at high risk of fracture in Japan treated with teriparatide 20 mcg daily for up to 24 months, included 33 osteoporotic patients with stage 4 or 5 CKD. In these patients, teriparatide was well tolerated and no new safety concerns were observed.[69]
Overall, however, we should state that the data on PTH administration in CKD are from very limited studies, including predominantly anecdotal data from patients who have low bone turnover, some of which are biopsied proven. Also, there is no information on the effect of intermittent PTH injections on phosphaturia or prevention of FGF-23 production. In addition, its safety should be tested in long-term prospective studies.
In all, it could be argued that intermittent PTH administration could trigger further amplifying feedbacks, upsetting further the disturbed Ca and Pi homeostasis. HPT might get worse and since it is associated with cardiovascular calcification and mortality, it is possible that PTH treatment could lead to similar adverse consequences. These are strong possibilities and should not be disregarded, until solid data suggest otherwise.
Go to :
CONCLUSIONS
Ca and Pi homeostasis is regulated by the interplay of PTH, 1,25D and FGF-23 (also cytokines and growth factors) through their action on bone, kidney and intestine. Renal impairment disturbs this balance and during progressive CKD, retention of Ca and Pi lead to the development of CKD-MBD. Currently, there is no effective treatment for this complication that includes a wide spectrum of different forms of metabolic bone diseases present in renal osteodystrophy, and vascular calcification. The most commonly used regimens are a range of Pi-lowering and anti-PTH agents.
The main argument for the use of PTH in the management of CKD-MBD is that the HPT that ensue is secondary. Until now, regardless how hard we try to compensate for that by manipulating the levels of Ca and Pi or 1,25(OH)2D, the other hormone affecting their homeostasis, our success is very limited.
The desirable scenario for early intermittent administration of exogenous PTH, a phosphaturic hormone, at a point in CKD (perhaps even stage II) when PTH is still normal, but the elevation of FGF-23 may be the signal that phosphorus retention is beginning, would be a mitigation of the development of MBD in CKD. More specifically, it could restore the homeostatic balance of phosphorus's impact on FGF-23 and also on PTH elevations, prevent the early decline of 1,25(OH)2D levels, and the proliferation/hyperplasia of the parathyroid glands. In addition, in contrast to catabolic effects of the continuously elevated PTH of the hyperparathyroid state, the intermittent administration of PTH could have anabolic effects. Even at the stage when endogenous PTH is raised, such an approached could be beneficial, provided that autonomous or tertiary HPT has not developed. Furthermore, vascular calcification could be prevented or slowed down significantly. Limited experimental and clinical data support such an outcome. Regarding its safety, any of the reported adverse effects could be milder because PTH could be administered, especially at the early stages of CKD-MBD, at a lower dose and at less frequent intervals compared to those we use for osteoporosis where the aim is to achieve positive bone balance. In the case of renal impairment, we seek to slightly supplement the PTH levels in order to restore the Ca and Pi homeostasis.
In the opposite scenario, negative effects could occur with PTH treatment. It could facilitate the development of HPT and accelerate vascular calcification. Also, the required dose might exceed the one used currently for the treatment of osteoporosis.
Go to :
 XML Download
XML Download