

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Oxidative stress plays an important role in the complex regulation of inflammation as well as in the pathogenesis of various chronic inflammatory diseases such as asthma.1 Many types of cellular stimuli, including antigens, infectious agents, and chemical mediators, induce a transient increase in intracellular reactive oxygen species (ROS).2 Asthmatic patients, in particular, exhibit increased oxidative stress, which is linked to bronchial hyperresponsiveness, excessive mucin secretion, and worsening airway inflammation.345 It was reported that the increased oxidative stress in asthmatic patients is related to lower pulmonary function.6
Although the precise mechanism of how increased oxidative stress is involved in asthmatic inflammation remains to be clarified, ROS is probably crucial to the modulation of immune cell responses through increased production of several pro-inflammatory cytokines and chemokines.7 Reduction-oxidation (redox) homeostasis is critical for maintaining normal cellular function and is tightly regulated by controlling the intracellular concentration of hydrogen peroxide through its generation via nicotinamide adenine dinucleotide phosphate oxidases and its scavenging via various antioxidant enzymes. Thus, intracellular antioxidants, such as peroxiredoxins, a pivotal member of the intracellular redox system, play a role in sustaining this homeostasis.89 Peroxiredoxins are a family of ubiquitous and abundant intracellular antioxidant enzymes with 6 isoforms identified to date.10 Among them, peroxiredoxin-6 is highly expressed in the lungs and acts as a scavenger to catalyze hydrogen peroxide.11
Oxidative stress can cause various modifications to intracellular components including DNA, proteins, lipids, and carbohydrates. Several post-translational modifications have been observed in intracellular proteins, resulting from direct oxidation or attachment of chemical groups by other cellular components oxidized to amino acid side chains.12 Recent studies have shown that ROS cause post-translational modifications including phosphorylation, acetylation, and oxidation in peroxiredoxins and these modifications play a major role in modulating peroxiredoxin activities.13 In addition, phosphorylated, acetylated, or oxidized proteins are selectively recognized and degraded by the proteasome system to minimize threats to survival.141516 The stability of redox-sensitive proteins is regulated through oxidative modifications, and the oxidized proteins are readily degraded by proteasomes and autophagy.17
In our previous study, we found that the ratios of hyperoxidized peroxiredoxins to total peroxiredoxins in peripheral blood mononuclear cells (PBMCs) were significantly higher in asthmatic patients than in healthy subjects and were correlated with disease severity as the highest ratio was seen in severe asthmatic patients.18 Based on these findings, we speculated that some dysregulation of the post-translational modification of peroxiredoxin-6 and intracellular control of its modified forms in bronchial epithelial cells (BECs) may be involved in the pathogenesis of asthmatic airway inflammation.
To date, modifications of peroxiredoxin-6 in the PBMCs of asthmatic patients have not been thoroughly investigated, nor has the relationship between the modification of peroxiredoxin-6 and down-regulation of its expression level. In this study, we examined the expression levels and post-translational modifications of peroxiredoxin-6 in the immune cells of asthmatic patients. We also investigated whether peroxiredoxin-6 is modified and whether its stability is altered by oxidative stress in BECs.
MATERIALS AND METHODS
PBMCs of study subjects
We recruited 22 asthmatic patients and 5 healthy individuals. All asthmatic patients were diagnosed by a physician (allergy specialist) and met criteria as defined in the Global Initiative for Asthma guidelines. The mean age, sex, and body mass index of the 2 groups were similar. Most of the patients were never smokers, and the average smoking amount was lower than 10 pack-years. The mean inhaled corticosteroids (ICS) budesonide equivalent doses were 510 mg/day, corresponding to the medium dose. The mean forced expiratory volume in 1 second (%) of patients was 75.64%. Most patients had chronic rhinosinusitis as a comorbid disease. The demographics of these patients are summarized in Table. Whole blood was drawn by clean venipuncture and collected into vacutainer tubes containing ethylenediaminetetraacetic acid (EDTA). PBMCs were isolated from whole blood under sterile conditions using Lymphoprep (Axis-Shield PoC AS, Oslo, Norway). Written informed consent was obtained from all participants prior to the study, and the study protocol was approved by the Institutional Review Board (approval number: 2013-0560).
Table
Baseline demographics of asthmatic patients
Cell culture
According to the recommendation by the American Type Culture Collection, BEAS-2B cells were grown in LHC-9 medium (Gibco, Grand Island, NY, USA) at 37°C in a CO2 incubator with a humidified atmosphere of 5% CO2 and 95% air.
Antibodies and reagents
Anti-peroxiredoxin-1, anti-peroxiredoxin-2, anti-peroxiredoxin-3, and anti-peroxiredoxin-SO3 were obtained from Abcam (Cambridge, MA, USA). Anti-peroxiredoxin-6 and β-actin were purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Anti-phosphothreonine and anti-acetylated lysine were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti-phosphoserine antibody was obtained from Thermo Fisher Scientific (Waltham, MA, USA). Horseradish peroxidase-conjugated secondary antibodies were purchased from Enzo Life Sciences (Farmingdale, NY, USA). Protein G-Sepharose and IPG buffer pH 4–7 were obtained from GE Healthcare Life Sciences (Pittsburgh, PA, USA). Hydrogen peroxide, MG132, and cycloheximide were purchased from Sigma Aldrich (St. Louis, MO, USA). Lactacystin was obtained from AG Scientific, Inc. (San Diego, CA, USA).
Expression patterns of peroxiredoxins in PBMCs
The expression patterns of the peroxiredoxin isoforms in PBMCs were assayed by Western blotting. Briefly, aliquots of PBMC lysates were loaded onto sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels and transferred to polyvinylidene difluoride (GE Healthcare Life Sciences) membrane. After blocking with 3% skim milk (BD Biosciences, San Diego, CA, USA) in TBS-T (20 mM Tris-HCl pH7.4, 150 mM NaCl, and 0.1% Tween 20), the immunoreactive proteins were detected with primary antibodies overnight at 4°C. The blots were subsequently incubated with horseradish peroxidase-conjugated secondary antibodies for 1 hour at room temperature, and the antibody-antigen complexes were detected using a chemiluminescence detection kit (ATTO Corporation, Tokyo, Japan). For the quantification of the expression levels of peroxiredoxin isoforms in the PBMCs lysates, chemiluminescence was detected using a LAS-4000 imaging system (Fujifilm, Tokyo, Japan) and analyzed using Multi Gauge version 2.2 software (Fujifilm).
Two-dimensional (2D) gel electrophoresis
Cell lysates were precipitated with TCA/acetone. Protein pellets were resuspended with rehydration buffer (7 M Urea, 2 M thiourea, 4% CHAPS, 2% DTT, 0.5% IPG buffer pH 4–7, 1X protease inhibitor cocktail [EMD Millipore Corp., Billerica, MA, USA], 1 mM Na3VO4). Afterward, 100 μg of each protein sample was loaded onto strip gels (7 cm, pH 4–7; GE Healthcare Life Sciences) and cover fluid (GE Healthcare Life Sciences) was added to the manifold cup-loading system. Samples were placed on IPGphor (GE Healthcare Life Sciences), rehydrated for 12 hours, and electrofocused. The separated strip gels were incubated in equilibration buffer (50 mM Tris-HCl pH 8.8, 6 M Urea, 2% SDS, 30% glycerol, 0.002% Bromophenol Blue) containing 10 mg/mL DTT for 15 minutes and then incubated with equilibration buffer containing 25 mg/mL iodoacetamide instead of DTT for 15 minutes. The equilibrated strip gels were applied to 1.0-mm thick 12% acrylamide gels and sealed with 0.8% (w/v) agarose solution. SDS-PAGE was carried out at 20 mA for 6 hours. The gels were stained by coomassie brilliant blue R-250 or assayed by Western blotting.
Immunoprecipitation analysis
BEAS-2B cells were treated with hydrogen peroxide in complete medium for the indicated time intervals. Cells were washed twice with ice-cold phosphate-buffered saline, lysed in lysis buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1X protease inhibitor cocktail, 1 mM Na3VO4) on ice for 10 minutes, and centrifuged at 21,124 g for 10 minutes at 4oC. The cell lysates were incubated with anti-peroxiredoxin-6 antibody at 4°C overnight, and then with protein G-Sepharose for 2 hours. The resin was washed 3 times in lysis buffer. Proteins bound to the resin were mixed with SDS sample buffer containing 10 mM β-mercaptoethanol, resolved by SDS-PAGE, and analyzed by Western blot. The levels of modifications analyzed using Multi Gauge version 2.2 software (Fujifilm).
Protein degradation assay
BEAS-2B cells were exposed to hydrogen peroxide in Hanks' balanced salt solution for the indicated times, washed, and incubated in complete medium containing the 40 μM cycloheximide with or without proteasome inhibitors (MG132, lactacystin) for the indicated times. The degradation rates of peroxiredoxin-6 in these cell lines were observed using Western blot analysis and analyzed using Multi Gauge version 2.2 software (Fujifilm).
Statistical analysis
All data are expressed as mean ± standard deviation of 3 independent experiments. Statistical analysis was performed using Microsoft Excel (version, 2007; Microsoft Corporation, Redmond, WA, USA). Differences between the groups were assessed using Student's t test. P < 0.05 was considered statistically significant.
RESULTS
Analysis of expression levels of peroxiredoxin isoforms in PBMCs from study subjects
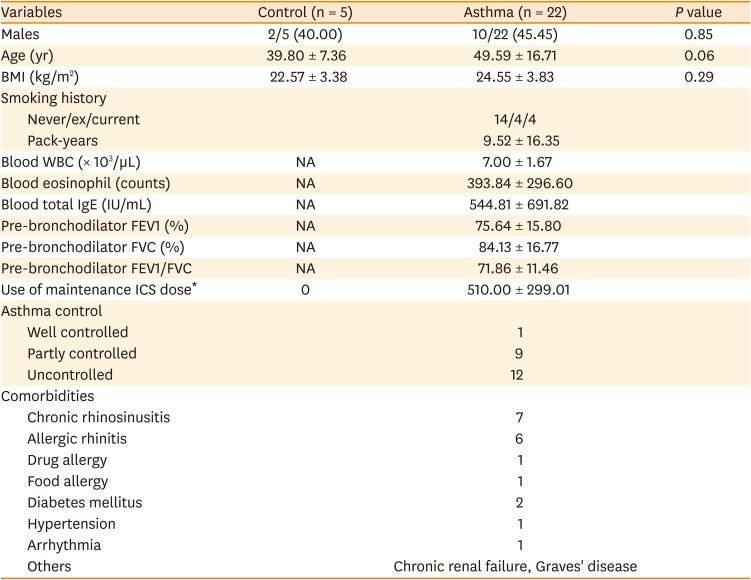
The expression levels of peroxiredoxin-1, 2, 3, and 6 in the PBMCs of asthmatic patients were analyzed by Western blotting. The clinical characteristics of asthmatic patients and healthy controls are summarized in Table. While the expression levels of peroxiredoxin-1, 2, or 3 showed no difference between the asthmatic patients and healthy controls, the expression of peroxiredoxin-6 in the PBMCs of asthmatic patients was consistently about 70% less on average than that of healthy controls (Fig. 1A and B, and Supplementary Fig. S1).
Fig. 1
Down-regulation and modification of Prdx6 in PBMCs of asthmatic patients. (A) PBMCs were prepared from each subject separately and various Prdxs were detected by immunoblotting. Lysates of PBMCs were separated on SDS-PAGE and detected with anti-Prdx6, 1, 2, and 3. β-actin was used as the loading control. Analysis was performed with 6 asthmatic patients and 5 healthy subjects and a single gel is presented as an example. (B) The quantified data from (A) are presented as a graph. The expression level of each Prdx in healthy control was set as 100%. (C) Representative data from 2D-PAGE gel on a non-linear pH (4–7) and Western blotting using anti-Prdx6 for PBMC lysates from 2 healthy subjects and 6 asthmatic patients.
Prdx, peroxiredoxin; PBMC, peripheral blood mononuclear cell; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis; 2D-PAGE, 2-dimensional-polyacrylamide gel electrophoresis; IEF, isoelectric focusing; IB, immunoblotting.
***P < 0.001 vs. healthy controls.
Peroxiredoxin-6 modification in PBMCs from asthmatic patients
Next, we determined whether peroxiredoxin-6 is post-translationally modified in the PBMCs of asthmatic patients. The protein spots for peroxiredoxin-6 in asthmatic patients were shifted to a more acidic region. Spots for peroxiredoxin-6 were located on spots 1 and 2 in healthy subjects, but those in asthmatic patients were located on spots 3 and 4 (Fig. 1C), suggesting that peroxiredoxin-6 is differentially and post-translationally modified in asthmatic patients.
Induction of peroxiredoxin-6 modification in BECs under oxidative stress
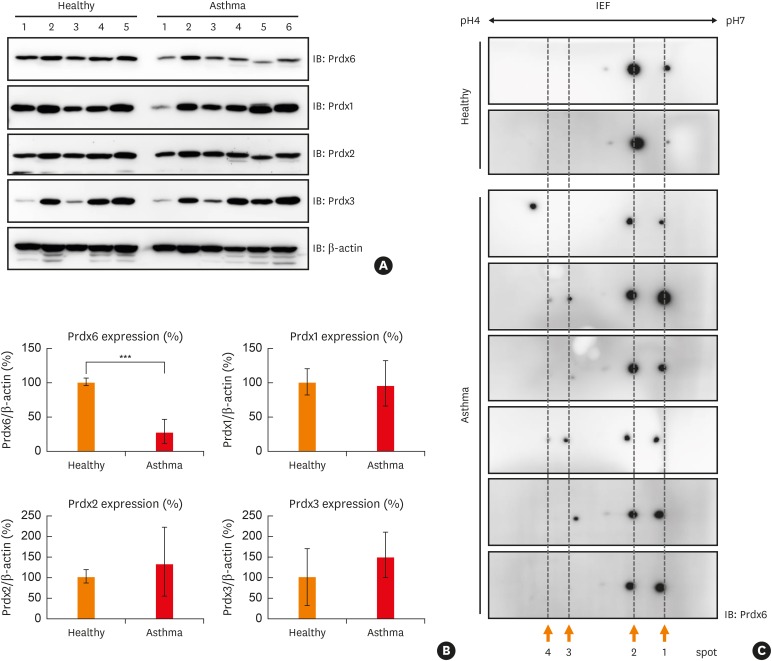
In order to investigate whether peroxiredoxin-6 modifications take place in BECs under oxidative stress, we first examined whether modifications of peroxiredoxin-6 are induced after treatment with hydrogen peroxide using BEAS-2B cells. Various proteins in lysates from BEAS-2B cells exposed with hydrogen peroxide were shift to acidic end of gels (Supplementary Fig. S2A). Spots for peroxiredoxin-6 were shifted to the left in hydrogen peroxide-treated cells compared to mock-treated cells (Fig. 2A). We observed that the levels of intracellular ROS in BECs exposed to hydrogen peroxide were rapidly increased after treatment for 10 minutes and decreased slowly until 3 hours (Supplementary Fig. S2B). The post-translational modifications of peroxiredoxin-6 were also detected in U937 cells (human macrophage cell line) and Jurkat cells (human T lymphocyte cell line), similar to BEAS-2B cells treated with hydrogen peroxide (Supplementary Fig. S3). In immunoprecipitation analysis, the levels of phosphorylated serine and threonine and acetylated lysine in the peroxiredoxin-6 in BEAS-2B cells exposed to different concentrations of hydrogen peroxide were increased in a dose-dependent manner (Fig. 2B and C). The oxidized peroxiredoxin-6 level was also increased (Fig. 2B and C).
Fig. 2
Various modifications of Prdx6 under oxidative stress. (A) Cell lysates from BEAS-2B cell line after mock, 25 μM or 50 μM H2O2 treatment for 3 hours were separated in 2D-PAGE gels and blotted to PVDF membrane. Prdx6 was detected with anti-Prdx6 antibody. (B) BEAS-2B cell lysates treated with mock, 25 μM, or 50 μM H2O2 for 3 hours were IP with anti-Prdx6 antibody. Phosphorylated serine or threonine residues, acetylation, and oxidation of Prdx6 were analyzed using the indicated antibodies. β-actin served as loading control. (C) The quantified data from (B) are presented as a graph. Each ratio of pSer, pThr, Ac-Lys, and oxidized Prdx6/Prdx6 in mock control was set as 100%. Values are the mean ± standard deviation (n = 3).
IP, immunoprecipitation; 2D-PAGE, 2-dimensional-polyacrylamide gel electrophoresis; PVDF, polyvinylidene difluoride; IEF, isoelectric focusing; IB, immunoblotting; Prdx, peroxiredoxin; H2O2, hydrogen peroxide.
*P < 0.05 vs. mock control.
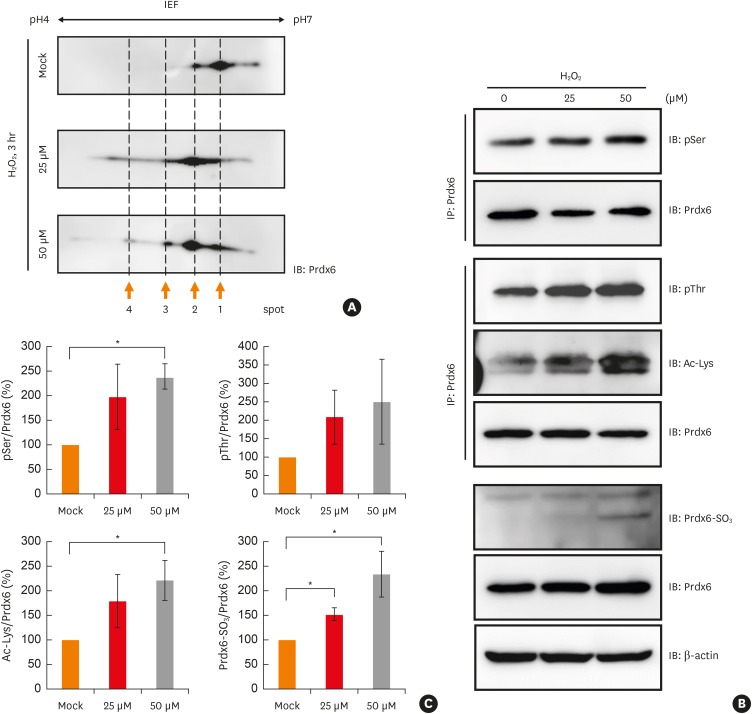
Modifications of peroxiredoxin-6 were analyzed in terms of the time course in BEAS-2B cells treated with different concentrations of hydrogen peroxide. Phosphorylation, acetylation, and oxidation of peroxiredoxin-6 increased in a dose- and time-dependent manner until 24 hours after hydrogen peroxide treatment, with an exceptional decrease in oxidized peroxiredoxin-6 (Fig. 3A and B).
Fig. 3
Analysis of oxidative stress-induced modifications Prdx6 in BEAS-2B cells. (A) BEAS-2B cells were treated with 25 μM or 50 μM hydrogen peroxide for indicated times. Phosphorylated serine or threonine residues, acetylation, and oxidation of Prdx6 were analyzed using the indicated antibodies. (B) The quantified data from (A) are presented as a graph. Each ratio of pSer, pThr, Ac-Lys, and oxidized Prdx6/Prdx6 in mock control was set as 100%. Values are the mean ± standard deviation (n = 3).
Prdx, peroxiredoxin; IB, immunoblotting; IP, immunoprecipitation.
*P < 0.05, **P < 0.01 vs. mock control.
Analysis of peroxiredoxin-6 stability in BECs exposed to oxidative stress
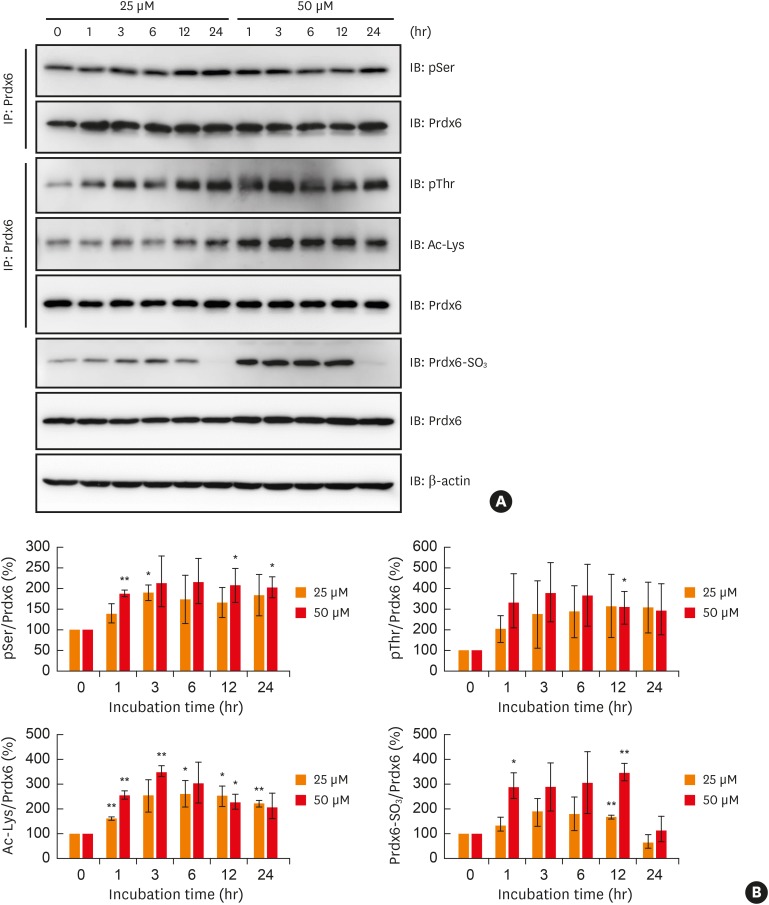
We evaluated whether the modifications to peroxiredoxin-6 under oxidative stress have an effect on its expression. We observed a significant reduction in peroxiredoxin-6 expression in hydrogen peroxide-treated BEAS-2B cells after incubation with cycloheximide, an inhibitor of protein synthesis, by immunoblotting (Fig. 4A and B). We analyzed the effect of oxidative stress on mRNA level of peroxiredoxin-6 in PBMCs from normal and asthmatic subject. The result showed that the levels of peroxiredoxin-6 gene transcripts were similar between the healthy and asthmatic patient and not change in both PBMCs exposed with hydrogen peroxide (Supplementary Fig. S4 and Supplementary Table).
Fig. 4
Degradation of Prdx6 modified by oxidative stress through proteasome in BEAS-2B cells. Degradation of Prdx6 through proteasome was examined by Western blot analysis. (A) BEAS-2B cells were treated with 50 μM H2O2 for 1 hour in HBSS. The cells were washed and incubated in complete medium containing 40 μM CHX for the indicated time. Cell lysates were separated on SDS-PAGE and detected with immunoblotting using anti-Prdx6 antibody. β-actin served as the loading control. (B) The quantified data from (A) are presented as a graph. The ratio of Prdx6/β-actin in mock control was set as 100%. Values are the mean ± SD (n = 3). (C) After 50 μM H2O2 treatment, BEAS-2B cells were incubated in 40 μM CHX with or without MG132 or LACT for 24 hours. (D) The quantified data from (C) are presented as a graph. The ratio of Prdx6/β-actin in BEAS-2B cells treated with H2O2 was set as 100%. Values are the mean ± SD (n = 3).
Prdx, peroxiredoxin; H2O2, hydrogen peroxide; SD, standard deviation; CHX, cycloheximide; HBSS, Hanks' balanced salt solution; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis; IB, immunoblotting; LACT, lactacystin.
*P < 0.05 vs. mock treatment for 24 hours; ††P < 0.01 vs. BEAS-2B cells treated with hydrogen peroxide; §§P < 0.01; §§§P < 0.001 vs. BEAS-2B cells treated with hydrogen peroxide and CHX.
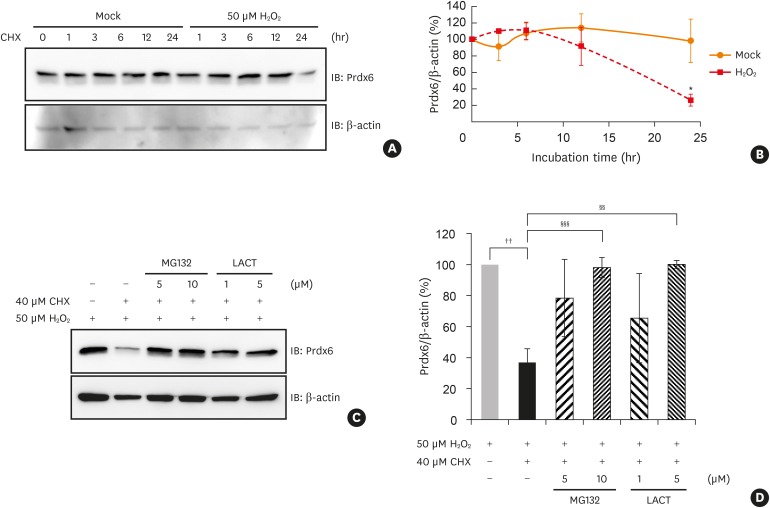
Next, we examined whether the peroxiredoxin-6 modified by oxidative stress would be degraded through a proteasome system. We found that the reduction of peroxiredoxin-6 expression under oxidative stress was blocked by treatment with MG132 or lactacystin, which are proteasome inhibitors (Fig. 4C and D).
DISCUSSION
In the current study, we found that intracellular peroxiredoxin-6 is post-translationally modified by phosphorylation and acetylation, and is significantly down-regulated in PBMCs from asthmatic patients and BECs exposed to oxidative stress as well. In addition, analysis suggested that the down-regulation of persoxiredoxin-6 probably resulted from proteasomal degradation of the modified forms. Given that peroxiredoxin-6 is abundant and plays a potential role in redox homeostasis in the lungs and airways, the down-regulation of peroxiredoxin-6 in immune cells and bronchial epithelium may have a substantial impact on the pathogenesis of asthmatic airway inflammation. Thus, it would be worth investigating the underlying mechanism of peroxiredoxin-6 reduction in cells under oxidative stress, which would further improve our understanding of asthma pathogenesis.
It is well known that oxidative stress in the airways is caused by various environmental stimuli such as allergens, pollutants, or endogenous ROS from inflammatory cells.19 Increased ROS levels in the airways leads to inflammation through cellular signaling cascades and was reported to be strongly related to the severity of asthma in patients.2021 Several studies have revealed that an imbalance between oxidants and antioxidants plays a significant role in the progression and severity of asthma.22 Furthermore, the dysfunction of these antioxidant activities is known as a characteristic of asthma.23 To maintain redox homeostasis, there are several enzymatic antioxidants such as superoxide dismutases (SODs), catalase, and glutathione peroxidases as well as heme oxygenase-1, thioredoxins, peroxiredoxins, and glutaredoxins in the lung tissues.24 In fact, it has been reported that the levels of antioxidants including glutathione peroxidase, SOD, reduced glutathione, ascorbic acid, α-tocopherol, lycopene, and β-carotene are significantly lower in asthmatic patients than those in healthy subjects.25 Other studies have reported that a defect in peroxiredoxin-2 is involved in the pathogenesis of asthma in a peroxiredoxin-2 knockout mice model.26 Additionally, liver injury is known to be increased by elevating hydrogen peroxide levels from mitochondria in peroxiredoxin-6-knockout mice.27
In the current study, therefore, we hypothesized that increased ROS levels in PBMCs and airway cells from asthmatic patients may induce various modifications to intracellular peroxiredoxin-6, one of the critical anti-oxidant molecules, and that the modifications could be associated with the dysregulation of redox homeostasis in the airway.
It is well known that both lung epithelial cells and immune cells are critically involved in the development of asthma and peroxiredoxin-6 is expressed in all major mammalian organs, with the highest expression levels in the lung tissues and lung epithelial cells. Furthermore, peroxiredoxin-6 is reported to play an important role in defense of the lung against oxidative stress.28 Based on those observations, we tried to evaluate if the findings from PBMCs of asthmatics are also meaningful in lung epithelial cells.
In our previous report, we stated that the amount of hyperoxidized peroxiredoxins was significantly higher in asthmatic patients than in healthy subjects.18 The levels of intracellular ROS in PBMCs of asthmatic patients exposed with hydrogen peroxide were significantly increased and remained higher compared with normal subject. In addition, we observed in the current study that among the peroxiredoxin isoforms, only peroxiredoxin-6 was reduced in PBMCs from asthmatic patients, but not in healthy controls. Taken together, it can be assumed that the decreased peroxiredoxin-6 in the asthmatic airway is associated with difficulties in maintaining redox homeostasis.
In the current study, we observed that various modifications to peroxiredoxin-6 were induced by oxidative stress in both the PBMCs of asthmatic patients and lung epithelial cells experimentally exposed to oxidative stress. It is widely known that peroxiredoxins can be regulated via various post-translational modifications including phosphorylation, acetylation, glutathionylation, and thiol oxidation.29 A recent study found that oxidized peroxiredoxin-6, which was examined by peptide sequencing, had various modifications including trioxidation and thiosulfonic acid at Cys47, acetylation at Lys63, and phosphorylations at Ser72, Ser146, Thr177, and Ser186.1730 Considering these findings, the oxidative stress-induced acidic shifts of peroxiredoxin-6 on 2D-electrophoresis observed in the current study can be explained by modifications such as oxidation, phosphorylation, and acetylation as well.
Our study revealed that peroxiredoxin-6 is highly modified and that its expression level is considerably decreased in asthmatic patients. This strongly suggests that the instability of the modified peroxiredoxin-6 is induced by oxidative stress. Since irreversibly oxidized proteins are an inevitable by-product of metabolism in mammalian cells, cells have developed diverse counteracting systems during evolution. These systems include some enzymes capable of repairing oxidative stress-induced damaged proteins, but their capacity is very limited. Consequently, cells need restorative systems to recognize and remove oxidatively modified proteins.31 For this purpose, protein quality control can resolve oxidative damage on proteins through the synthesis of new proteins, folding and refolding of oxidized proteins, and removal by proteolytic systems.17 In the event that the concentration of hydrogen peroxide increases above levels that can be controlled by cellular antioxidant enzymes, cells become damaged. Under such conditions, cells are likely to undergo apoptosis by targeting phosphorylated catalase for proteasomal degradation and thereby rapidly elevating hydrogen peroxide levels.32 We observed that oxidatively modified peroxiredoxin-6 was decreased by degradation through the proteasome system in BECs. These results indicated that the modification of peroxiredoxin-6 induced by oxidative stress may cause its degradation through the proteasome system. In the current study, the expression level of peroxiredoxin-6 in bronchoalveolar lavage fluid cell from asthmatic patients was not measured, which is a limitation of the present study.
Despite the results of increased oxidative stress levels and decreased peroxiredoxin-6 expression in PBMCs and BECs we presented in this study, there is no direct evidence of functional changes of modified peroxiredoxin-6. Unfortunately, we did not perform the experiment to see a direct effect of modified form of peroxiredoxin-6 on oxidative stress status in the current study. However, we provided a strong correlation between increased intracellular ROS and increased modification of peroxiredoxin-6 and decreased expression of peroxiredoxin-6 in hydrogen peroxide exposed cells. Thus, it can be assumed that increased modified peroxiredoxin-6 and decreased total perxoredoxin-6 may be linked to increased intracellular oxidative stress status. It is speculated that increased oxidative stress in asthmatic airway and immune cells, which is well known phenomenon, can critically associated with the pathogenesis of asthmatic airway inflammation through down-regulation of peroxiredoxin-6 leading to decreased ability to quench ROS and increased vulnerability to external oxidative stress load. Further studies on the precise role of modified peroxiredoxin-6 are warranted.
To date, whether the peroxiredoxin isoforms are reduced by oxidative modifications in inflammatory diseases such as asthma remains unclear. Herein, we demonstrated that peroxiredoxin-6 is highly modified and its expression is significantly reduced by oxidative stress in asthmatic patients. We also propose that this down-regulation of peroxiredoxin-6 may be associated with degradation of the modified form through proteasomes, resulting in poorer control of intracellular hydrogen peroxide levels. Finally, the results of the current study strongly suggest that the dysregulation of intracellular peroxiredoxin-6 may critically cause an imbalance between oxidants and antioxidants in asthmatic airway inflammation and may be one of the principle mechanisms underlying asthma pathogenesis. In addition, down-regulation and various modifications of peroxiredoxin-6 may serve as a novel biomarker for severe asthmatic patients. It might be possible to develop new approaches to treating asthma by restoring antioxidants such as peroxiredoxin-6, modulating the redox homeostasis.
 XML Download
XML Download