

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Chronic rhinosinusitis with nasal polyps (CRSwNPs) is a multifactorial and highly heterogeneous disease with persistent inflammation and remodeling of nasal mucosal tissue.1 Cytokine profiles of CRSwNP are diverse, based on the endotype, comorbidity, region, and race.23 Together with Th2 immune response, Th17 polarizations are increased in airway inflammatory diseases such as CRSwNP.456 Interleukin (IL)-17A, a cytokine produced mainly by Th17 cells, plays critical roles in the development of allergic reaction, inflammation, and autoimmunity.789
IL-17A is a major inflammatory molecule in psoriasis, and many clinical trials with anti-IL-17A monoclonal antibodies had shown therapeutic effects on it; therefore, the US Food and Drug Administration approved secukinumab for the treatment of psoriasis in 2015.101112 In asthma, which is mainly driven by Th2-mediated inflammatory processes, Th17-related inflammation plays a crucial role in patients with severe type, steroid-resistance, and acute exacerbation.131415 Brodalumab, an anti-IL-17A receptor monoclonal antibody, was randomly administered to patients with moderate to severe asthma for phase 2 clinical trial, and clinical effects were observed only in the subgroup with high bronchodilator reversibility, rather than in all study participants.16 Although clinical trials with anti-IL-17A monoclonal antibodies have not been conducted in CRSwNP so far, previous articles had reported IL-17A to be overexpressed in nasal polyps (NPs) and be associated with tissue remodeling, eosinophilic accumulation, and neutrophilic infiltration.171819
Blockage of mechanistic target of rapamycin (mTOR) exhibited a protective effect on inflammatory diseases.2021 Previous articles had reported mTOR-positive inflammatory cells to be increased in NP tissues, and activation of mTOR (phosphorylated mTOR, p-mTOR) in NP to be associated with regulatory T-cell insufficiency and autophagy deficiency that could affect the pathogenesis of CRSwNP.2223
The cytokine network of Th17-associated mediators in CRSwNP, including IL-17A, IL-23, and tumor necrosis factor (TNF)-α, is not fully understood yet. In this study, we aimed to identify the role of IL-17A in CRSwNP and to analyze the source of IL-17A-producing cells in NP. We also evaluated the effects of IL-17A and TNF-α blockage on NP formation in a murine NP model. Moreover, we sought to investigate whether the inhibition of mTOR signal pathway could suppress IL-17A expression and NP formation in the mouse NP model.
MATERIALS AND METHODS
Patients and tissue samples
Sinonasal tissues were harvested from subjects with healthy control, CRSwNP, and chronic rhinosinusitis without nasal polyp (CRSsNP). The diagnosis of CRSwNP was established based on the European position paper on rhinosinusitis and NPs 2012 guideline.24 Exclusion criteria were as follows: 1) younger than 18 years of age; 2) asthmatic or sensitive to aspirin; 3) prior treatment with antibiotics, systemic or topical corticosteroids, or other immune-modulating drugs for 4 weeks before surgery; and 4) combined with other nasal diseases including antrochoanal polyp, allergic fungal sinusitis, cystic fibrosis, or immotile ciliary disease. Control tissues were obtained from patients without any sinus diseases with negative skin prick test response during rhinologic surgeries. Uncinate process (UP) tissues were collected from patients with CRSsNP and control subjects, and UP and NP tissues were collected from patients with CRSwNP. The study was approved by The Institutional Review Board (IRB No. 2012-11-008-002) and informed consent was obtained from all patients. Demographic data are presented in Table.
Table
Demographics of subjects and study methodologies
Data are expressed as mean ± standard deviation or number of patients (%).
CRSsNP, chronic rhinosinusitis without nasal polyp; CRSwNP, chronic rhinosinusitis with nasal polyp; Ig, immunoglobulin; IHC, immunohistochemistry; IF, immunofluorescence.
*A P value < 0.05 was considered statistically significant for all analyses.
![]()
Immunohistochemistry (IHC)
IHC was conducted using Avidin-Biotinylated-Horseradish Peroxidase kits (Vector Laboratories, Burlingame, CA, USA) and DAB-Detection System (Golden Bridge International Labs, Bothell, WA, USA). After deparaffinization, the sections were rehydrated and boiled at 121°C for 10 minutes in 100 mM citrate buffer (pH 6.0) (Dako, Santa Clara, CA, USA) for heat-induced epitope retrieval. The sections were treated and incubated with primary antibodies and biotin-conjugated secondary antibodies. We used primary antibodies for human sinonasal tissues with IL-17A (1:200, rabbit immunoglobulin [Ig]G, ab136668; Abcam, Cambridge, UK), IL-23 (1:200, rabbit IgG, ab115759; Abcam), TNF-α (1:100, rabbit IgG, ab6671; Abcam), and p-mTOR (1:100, rabbit IgG, 2976; Cell Signaling Technology, Danvers, MA, USA). Mouse mucosal tissues were stained using primary antibodies such as IL-17A (1:50, rat IgG1, LS-B4912; LSBio, Seattle, WA, USA), CD68 (1:50, mouse IgG1k, MA5-13324; Invitrogen, Carlsbad, CA, USA), and Neutrophil (NIMP-R14, 1:50, rat IgG2b, ab2557; Abcam). No primary antibody control and/or isotype control were used for reagent control.
Confocal microscopy
To investigate the co-localization of IL-17A and IL-23, double immunofluorescence (IF) was performed using IL-17A antibody (1:50, goat IgG, AF-317; R&D Systems, Minneapolis, MN, USA) and IL-23 antibody (1:200, rabbit IgG, ab115759; Abcam), respectively. Dual IF staining was performed to identify IL-17A (1:200, rabbit IgG; Abcam) expressing cells using cell markers including CD4 (1:50, mouse IgG1, MAB379, R&D Systems) for T helper cells, CD11c (1:100, mouse IgG1, ab11029; Abcam) for dendritic cells, CD56 (1:100, mouse IgG1, 3576, Cell Signaling Technology) for natural killer (NK) cells, CD68 (1:50, mouse IgG1k, MA5-13324; Invitrogen) for M1 macrophage, CD163 (1:100, mouse IgG1, ab156769; Abcam) for M2 macrophage, and human neutrophil elastase (ELA2, 1:50, mouse IgG1, MAB91671; R&D Systems) for neutrophils. After washing step, secondary antibodies were treated: Alexa Fluor 488 (1:200, Life Technologies, Carlsbad, CA, USA), biotinylated anti-rat (1:100, Vector Laboratories, Burlingame, CA, USA) and streptavidin-Cy3 (1:100, Sigma-Aldrich, St. Louis, MO, USA).
Experimental protocol of the murine NP model
Detailed experimental protocol of the murine NP model was described in a previous article.25 In brief, BALB/c mice (female, 13.5-15.5 g, 4 weeks of age) were divided into 5 groups (n = 8 for each group): 1) negative control, 2) positive control, 3) IL-17A neutralizing antibody (secukinumab, Cosentyx®; Novartis, Basel, Switzerland) treatment, 4) TNF-α neutralizing antibody (infliximab, Remicade®; Janssen, Beerse, Belgium) treatment, and 5) dexamethasone treatment. Another experiment was conducted using rapamycin and compared with negative control and positive control group (n = 8 for each group). Mice were injected with 25 µg of ovalbumin (OVA; Sigma-Aldrich) in 2 mg of aluminum hydroxide gel intraperitoneally on days 0 and 5, followed by a daily intranasal instillation with 100 µg OVA diluted in 20 µL of PBS from days 12-19. In addition, they were weekly challenged with 10 ng of Staphylococcus aureus enterotoxin B (List Biologic Laboratories, Campbell, CA, USA) from 5 through 12 weeks after OVA instillation. Anti-IL-17A antibody (20 mg/kg), anti-TNF-α antibody (5 mg/kg), rapamycin (3 mg/kg), and dexamethasone (1 mg/kg) were administered weekly intraperitoneally from day 49 through day 102 before OVA instillation. Mice in the negative control group was challenged with phosphate-buffered saline. Mice were killed on day 104. The committee on the use and care of animal approved all animal experiments and we followed strict governmental and international guidelines on animal experimentation (No. DKU-17-019).
Histopathologic evaluation and IHC of the murine NP model
Polypoid lesions in our murine NP model were defined as distinct mucosal elevations with eosinophilic infiltration and microcavity formation. The number of polypoid lesions were enumerated by 2 independent examiners using hematoxylin and eosin (H&E) stained mucosal samples. Samples were also stained to compare specific characteristics using IHC. Epithelial changes in mouse mucosal tissues were analyzed on H&E slide and were defined as the ratio of inflamed mucosal tissue length and total epithelial length in the same location of each group (Supplementary Fig. S1A). The numbers of positive cells in the subepithelial layer using IHC were counted in the densest tissue region in 5 high-power fields (HPF; ×400 magnification) by 2 independent observers and average values were scored.
Quantitative real-time polymerase chain reaction (PCR) analysis of the nasal mucosa cytokine profiles
Total RNA was prepared from the nasal mucosa with a TriZol reagent (Invitrogen). Complementary DNA (cDNA) was synthesized using Superscript reverse transcriptase (Invitrogen) and oligo(dT) primers (Fermentas, Burlington, Canada). For the analysis of IL-4 (Mm00445258_g1), IL-5 (Mm00439646_m1), interferon-γ (Mm99999071_m1), IL-17A (Mm0439618_m1), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Mm03302249_g1), Pre-Developed Assay Reagent kits of primers and probes were purchased from Applied Biosystems (Foster City, CA, USA). Amplification of cDNA was carried out on MicroAmp optical 96-well reaction plates (Applied Biosystems). The reaction was performed using a StepOnePlus™ real-time PCR System (Applied Biosystems). The average transcript levels of the genes were then normalized to GAPDH.
Measurement of IgE, IgG1, and IgG2a in mice serum
Serum levels of total IgE and OVA-specific IgE were quantified using solid-phase enzyme-linked immunosorbent assay. OVA-specific IgG1 and IgG2a were analyzed using biotinylated rat anti-mouse IgG1 (BD Pharmingen, San Jose, CA, USA) and IgG2a (BD Pharmingen), respectively.
Statistical analysis
Data were analyzed by the 1-way analysis of variance test with post hoc Tukey test for continuous variables and by the χ2 test or Fishers exact test for categorical variables, respectively. Correlations were tested by Pearson's correlation coefficients to assess the association between molecular markers. A P value of less than 0.05 was considered statistically significant. Analyses were performed using Stata software v14.0 (StataCorp LP, College Station, TX, USA) and Graphpad prism software 7.0 (GraphPad Software Inc., La Jolla, CA, USA).
RESULTS
Up-regulated IL-17A in CRSwNP
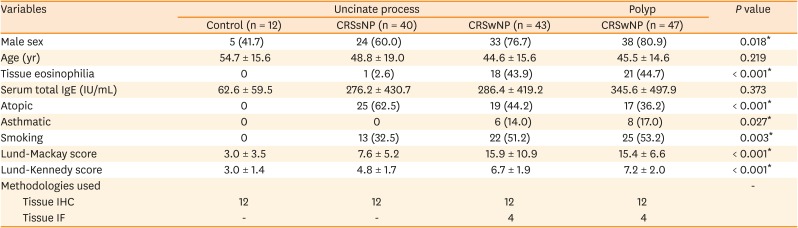
IHC showed the number of IL-17A+ inflammatory cells to be significantly increased in NPs from patients with CRSwNP compared to that in UP tissues from control subjects and patients with CRSsNP or CRSwNP (Fig. 1A). Moreover, UP tissues from patients with CRSsNP or CRSwNP exhibited higher proportion of IL-17A+ cells than those from control subjects. Expression of IL-23, TNF-α, and p-mTOR was also increased in NP tissues (Fig. 1B-D). While the number of IL-17A+ cells was significantly correlated with that of IL-23+ (P = 0.0122, r = 0.3191) and p-mTOR+ cells (P = 0.0473, r = 0.2791), no such correlation was seen between IL-17A+ cells and TNF-α+ cells (Fig. 1E). Upon double IF staining, some inflammatory cells exhibited colocalization of IL-17A and IL-23, suggesting their possible association (Fig. 1F).
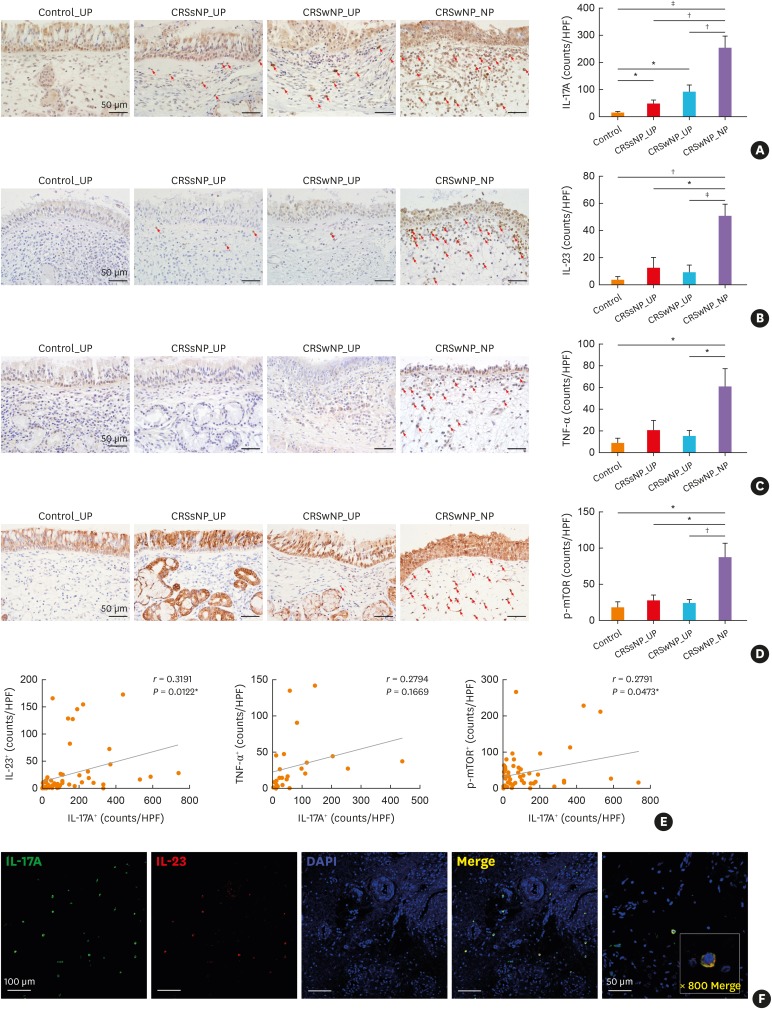
Fig. 1
Expression of IL-17A-associated inflammatory markers in human sinonasal tissues. (A and B) The number of IL-17A+ and IL-23+ cells (red arrows) according to the different types of tissues. (C) Expression of TNF-α. (D) Expression of phosphorylated-mTOR. (E) Correlation analysis of the number of IL-17A-positive cells with that of IL-23, TNF-α, and mTOR-positive cells in all human sinonasal tissues (n = 48, n = 24 for TNF-α). (F) Dual immunofluorescent staining for IL-17A and IL-23.
IL, interleukin; TNF, tumor necrosis factor; mTOR, mechanistic target of rapamycin; CRSsNP, chronic rhinosinusitis without nasal polyp; CRSwNP, chronic rhinosinusitis with nasal polyp; UP, uncinate process; NP, nasal polyp.
*P < 0.05, †P < 0.01, and ‡P < 0.001.
![]()
Cellular source of IL-17A in CRSwNP
To identify the cellular source of IL-17A in NP tissues, double IF staining with IL-17A antibody and various hematopoietic cell-specific markers was performed (Fig. 2A). We counted the colocalized cells, with IL-17A and each cell marker, using a confocal microscope. CD68+ M1 macrophages dominantly expressed IL-17A, followed by elastase+ neutrophils and CD4+ T helper cells (Fig. 2B). CD56+ NK cells, CD11c+ dendritic cells, and CD163+ M2 macrophages exhibited scanty colocalization with IL-17A in NPs.

Fig. 2
Dual immunofluorescent staining for each cell marker. (A) Double positive cells (white arrow) of IL-17A with CD68, ELA2 (elastase), CD4, CD56, CD11c, and CD163. (B) Total number of IL-17A-double positive cells.
IL, interleukin; DAPI, 4′,6-diamidino-2-phenylindole.
*P < 0.05, †P < 0.01, and ‡P < 0.001.
![]()
Neutralization of IL-17A and TNF-α reduces polyp formation and inflammation in murine NP model
To evaluate the role of IL-17A and TNF- α, we examined the effect of neutralizing antibodies of IL-17A and TNF-α using a previously established murine NP model (Fig. 3A).26 Neutralization of IL-17A and TNF-α effectively reduced the number of NPs (Fig. 3B) compared to that in the positive control group. Both neutralizing antibodies reduced NP formation as effectively as dexamethasone treatment. IL-17A+ cells were significantly reduced in number after treatment with anti-IL-17A antibody and dexamethasone while anti-TNF-α treatment tended to decrease IL-17A+ cell number without statistical significance (Fig. 3C). CD68+ cells, M1 macrophages that formed the main source of IL-17A, were effectively decreased in the 3 treatment groups compared to the positive control group (Fig. 3D). Neutrophil infiltration exhibited the same pattern as in CD68+ cells, whereas eosinophilic infiltration was only decreased in the dexamethasone treatment group (Fig. 3E and F). The percentage of epithelial change representing epithelial to mesenchymal transition (EMT) was increased in the positive control group compared to the negative control group. Percentage of EMT was reduced after treatment with anti-IL17 antibody, anti-TNF-α antibody, and dexamethasone (Supplementary Fig. S1B). Inflammatory cytokine levels of sinonasal mucosa, including IL-17A, IL-4, and IL-5, were significantly decreased in the 3 treatment groups (Supplementary Fig. S1C). Additionally, serum total IgE level was decreased following treatment with anti-IL17 antibody, anti-TNF-α antibody, and dexamethasone, whereas OVA-specific IgE level was decreased following dexamethasone treatment (Supplementary Fig. S1D).
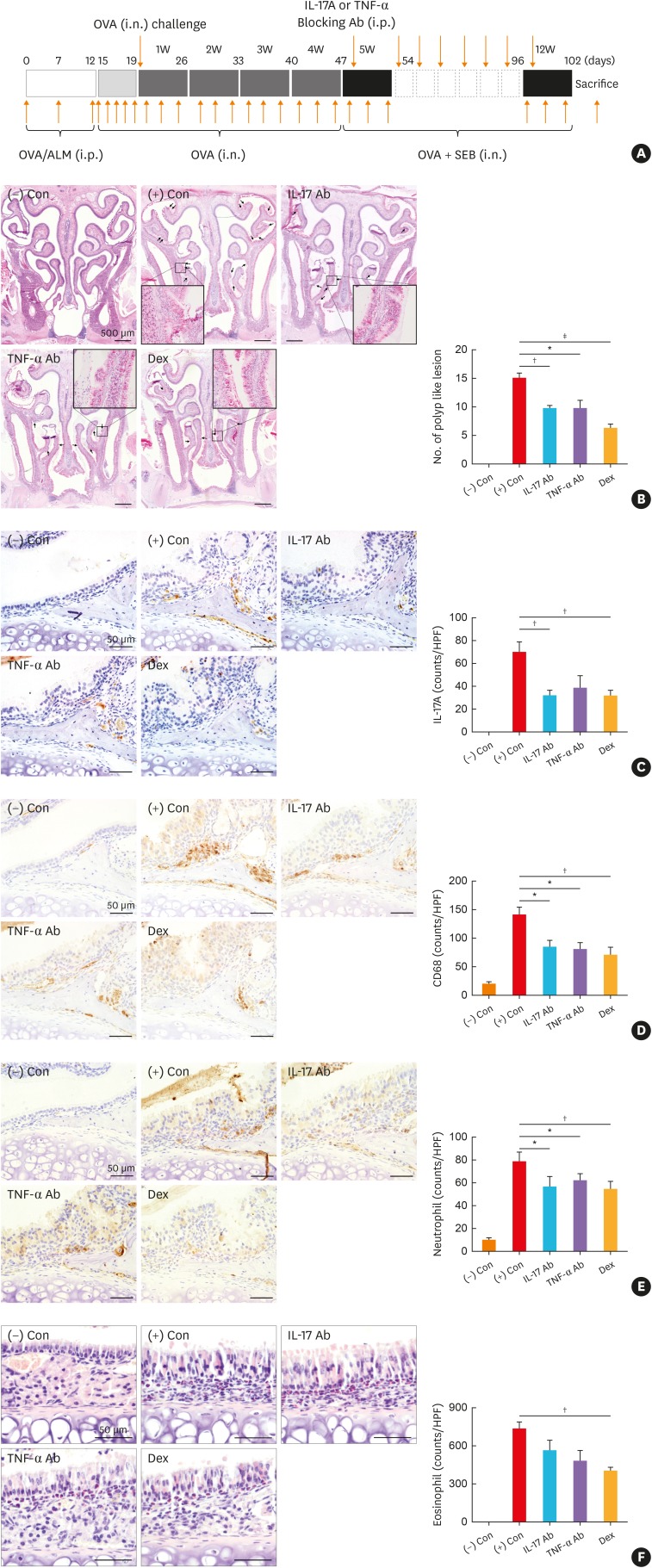
Fig. 3
Effect of anti-IL-17A and anti-TNF-α in the murine model. (A) Protocol of the murine model of chronic rhinosinusitis with nasal polyp. OVA and SEB were instilled into the nasal cavity to induce nasal polyposis. Anti-IL-17A and anti-TNF-α were administered intraperitoneally. (B) Number of polypoid lesions. (C and D) IL-17A+ and CD68+ cell counts. (E and F) Number of infiltrated neutrophils and eosinophils.
Subjects were divided into 5 groups; (−) Con: negative control group, (+) Con: positive control (nasal polyp) group, IL-17 Ab: IL-17 neutralizing antibody treatment group, TNF-α Ab: TNF-α neutralizing antibody treatment group, and Dex: dexamethasone treatment (therapeutic control) group.
OVA, ovalbumin; IL, interleukin; TNF, tumor necrosis factor; ALM, aluminum hydroxide; SEB, Staphylococcus aureus enterotoxin B; i.p., intraperitoneal; i.n., intranasal.
*P < 0.05, †P < 0.01, and ‡P < 0.001.
![]()
Inhibition of mTOR pathway using rapamycin suppresses polyp formation and inflammation in murine NP model
The mTOR pathway is known to be related to the IL-17A signaling pathway, and its inhibition using rapamycin has been reported to result in the inhibition of IL-17. As an alternative way to evaluate the effect of IL-17 inhibition, mice were treated with rapamycin intraperitoneally (Fig. 4A). Results showed that rapamycin treatment in murine NP model effectively reduced the number of polyps (Fig. 4B), and IL-17A+ cell numbers were significantly reduced compared to that in the positive control group (Fig. 4C). CD68+ M1 macrophages and neutrophils, the main source of IL-17A, were also reduced in number following treatment with rapamycin (Fig. 4D and E). Furthermore, eosinophilic infiltrations in the sinonasal tissues and cervical lymph nodes were suppressed in the rapamycin treatment group compared to the positive control group (Supplementary Fig. S2A and S2B). EMT was restored after rapamycin treatment (Supplementary Fig. S2C). The mRNA levels of IL-4, IL-5, and IL-17A were significantly reduced in the rapamycin-treated group (Supplementary Fig. S2D) and serum levels of total IgE, OVA-specific IgG1, and IgG2a were also reduced following rapamycin treatment (Supplementary Fig. S2E).
Fig. 4
Effect of blocking mTOR pathway using rapamycin in the murine model. (A) Protocol of the murine model and rapamycin was administered intraperitoneally. (B) Number of polypoid lesions. (C and D) IL-17A+ and CD68+ cell counts. (E) Number of elastase+ neutrophils.
Subjects were divided into 3 groups; (−) Con: negative control group, (+) Con, positive control (nasal polyp) group, and Rapamycin: rapamycin treatment group.
mTOR, mechanistic target of rapamycin; IL, interleukin; OVA, ovalbumin; i.n., intranasal; i.p., intraperitoneal; ALM, aluminum hydroxide; SEB, Staphylococcus aureus enterotoxin B.
*P < 0.01, and †P < 0.001.
![]()
DISCUSSION
In this study, we identified Th17-associated inflammation to be up-regulated in NP tissues from patients with CRSwNP, CD68+ M1 macrophages being the main cellular source of IL-17A cytokine. Neutralization of IL-17A and TNF-α effectively reduced the number of NPs as well as the inflammatory burden in a murine NP model. This anti-inflammatory effect was also observed in the rapamycin treated group, where the mTOR pathway was block.
Previous studies on the cellular source of IL-17A in CRSwNPs have reported many types of inflammatory cells expressing IL-17A. In NP tissues from the Chinese population, 25% of IL-17A+ cells were neutrophils as per IHC study,27 whereas CD8+ cytotoxic T cells constituted the major source of IL-17A in flow cytometric analysis.20 Other cell types, including macrophages, T helper cells, NK cells, γδ T cells, and eosinophils, have been reported to produce IL-17A in CRSwNP.171828 However, controversies still remain regarding the main cellular source of IL-17A, not only in CRSwNP but also in other inflammatory diseases. IL-17A is produced by innate and adaptive immune cells during inflammation or infection.29 A previous study have demonstrated eosinophilic chronic rhinosinusitis (CRS) to exhibit higher expression of IL-17+ cells compared to non-eosinophilic CRS, a significant correlation was seen between the number of IL-17A+ cells and CD68+ macrophages.30 In a mouse model of asthma, IL-17A induced an accumulation of macrophages along with their prolonged survival.31 In this study, CD68+ M1 macrophages were the main cellular source of IL-17A in NP tissues. Almost 60% of CD68+ M1 macrophages expressed IL-17A, whereas CD163+ M2 macrophages revealed scanty co-expression of IL-17A. These results are consistent with the previous report that revealed M1 macrophages to induce pro-inflammatory cytokines and Th1 inflammatory responses, while M2 macrophages were attributed to produce eosinophilic cytokines in the pathogenesis of CRSwNP.32
Wang et al.33 recently published about crosstalk between Th2 and Th17 inflammation in CRSwNP. Human dispersed NP cells were stimulated with Th2 or Th17 cytokines. The result was that Th17 cytokines increased levels of Th2 cytokines, which implies Th2 inflammation is enhanced by Th17 stimulation in CRSwNP. Another study has investigated that epithelial derived cytokines, such as IL-1β, IL-23, and TGF-β, can differentiate group 2 innate lymphoid cells (ILCs) into IL-17-producing ILCs in nasal inflammation.34 Th17 inflammation is not a main player of polypogenesis, whereas it can enhance or aggravate Th2-associated inflammatory responses. The specific signaling pathways between Th2 and Th17 inflammation need to be investigated in the further research.
A previous paper showed the contradictory results to our results.35 It showed that the number of polypoid lesions in a murine NP model was not significantly different between wildtype and IL-17A knockout mice. However, in the paper, IL-17A knockout mice had significant reduction in eosinophils and neutrophils infiltration, subepithelial fibrosis, and goblet cell count. It suggests reduced inflammation and remodeling, which can lead to a possible reduction of polypoid lesions. If more polypoid lesions developed in both mice groups, the results would have reached statistical significance.
TNF-α and IL-17A have been shown to exert synergistic effects in psoriasis, one of the chronic inflammatory skin diseases.3637 TNF-α stimulates dendritic cells to produce Th17-associated cytokines, and its inhibition reduces the proliferation of dendritic cells and Th17 cells along with the inflammatory molecules produced.3839 In this study, TNF-α-blocking antibody treatment ameliorated inflammation in NP model as effectively as anti-IL-17A-blocking antibody or dexamethasone treatment. Clinically, low-dose long-term macrolide therapy has been reported to exert benefit to non-eosinophilic CRS via blockage of pro-inflammatory cytokines, particularly TNF-α.4041 Hence, anti-TNF-α treatment might be a therapeutic strategy in some specific endotypes of CRSwNP and could be administered in combination with anti-IL-17 blocking antibody.
Activation of the mTOR pathway is well known to enhance Th17 cell differentiation through several downstream mechanisms such as hypoxia-inducible factor (HIF)-1α or signal transducer and activator of the transcription (STAT) pathway.42 Both HIF-1α and STATs have already been identified as pathogenic mechanisms of NPs.4344 Inhibition of mTOR pathway using rapamycin has been studied for its anti-inflammatory effects on various diseases.4546 In CRSwNP, activation of the mTOR pathway is associated with regulatory T-cell insufficiency, and inhibition of mTOR using rapamycin reduces myofibroblast differentiation, extracellular matrix production, and collagen deposition.2247 Our results of this animal experiment demonstrated that rapamycin treatment attenuates the expression of IL-17A by decreasing the number of IL-17A-producing cells. Although the anti-inflammatory effect of rapamycin was not directly compared to that of neutralizing antibody or dexamethasone treatment, rapamycin treatment was found to significantly reduce inflammatory burdens in the murine NP model. Although mTOR is expressed in many cell types, it is not fully studied which specific cells are expressed in NP tissues. Regulatory T cells were enhanced in cultured NP tissues after inhibition of mTOR signaling pathway.22 In a colitis model, blocking of the mTOR pathway inhibits production of IL-17, IL-22, and IL-23 in neutrophils.48 It can be another possible therapeutic mechanism in NPs.4950
One limitation of the present study is the lack of mechanistic experiment, including in vitro testing, to prove a molecular association among IL-17A, TNF-α, and the mTOR pathway. Further research is warranted to investigate a cellular mechanism of IL-17A and the mTOR signaling pathway in nasal polypogenesis. Additionally, subgroup analysis between eosinophilic and non-eosinophilic CRS would be required to confirm the anti-inflammatory effect of mTOR inhibition. However, our study revealed that IL-17A may play a crucial role in the pathogenesis of CRSwNP, whose major cellular source in human NP tissues is M1 macrophage. Targeting IL-17A directly or indirectly might be a good therapeutic approach in CRSwNP.
 XML Download
XML Download