

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
The airways are organs that are constantly exposed to airborne pathogens and foreign antigens throughout life. Therefore, various kinds of cells in the airways, including structural cells and immune cells, interact to form a precise defense system against pathogens and foreign antigens that involves both innate immunity and acquired immunity. In recent years, innate lymphoid cells (ILCs), which are widely conserved in humans and non-human vertebrates, such as the mouse and zebra fish, have attracted attention as cells involved in inflammatory diseases as well as defense against pathogens and foreign antigens.
Although ILCs have a lymphoid morphology, unlike T cells and B cells they do not have antigen receptors that recognize the microstructures of antigens; thus, they are not directly activated by specific antigens. They are activated mainly by antigen non-specific mechanisms, suggesting that they are involved in innate immunity. Some ILCs express toll-like receptors (TLRs) that recognize microbes, and the cells may be directly activated by the pathogen-associated molecular patterns of microbes. However, some recent articles have shown that ILCs express various kinds of receptors for cytokines, danger signals, neuropeptides and lipid mediators that are more dominant than TLRs. These findings suggest that ILCs may be activated by signals from other cells around it upon exposure to foreign antigens (including microbes), rather than by being directly activated by foreign antigens.
ILCs exhibit substantial heterogeneity, with different phenotypes and functions depending on the organ and type of inflammation, presumably because of differences in microenvironments. Thus, ILCs may be a sensitive detector of microenvironmental changes at our body surfaces, such as mucosal sites and the skin. Conversely, by analyzing the phenotype and function of ILCs, we can better understand the details of the microenvironment at local sites in airway diseases. In this review, we aimed to identify molecules that either positively or negatively influence the function and/or plasticity of ILCs and the sources of molecules in the airways in order to examine the pathophysiology of airway inflammatory diseases and facilitate the issues to be solved.
Go to : 
ILC SUBSETS AND THEIR PLASTICITY
ILCs have a lymphoid morphology, but they have no cell-surface lineage markers such as those that define other immune cell subsets. Instead, all ILCs express a cytokine receptor subunit called interleukin (IL)-7 receptorα (CD127). ILCs are currently divided into 3 different subtypes, according to their expression of cytokines and transcription factors: group 1 ILCs (ILC1s), group 2 ILCs (ILC2s) and group 3 ILCs (ILC3s). ILC1s are defined as ILCs that express T-box-expressed-in-T cells (T-bet) and produce interferon (IFN)-γ; they include conventional natural killer cells (cNK) and are considered to be involved in anti-viral immunity, like T helper (Th)1 cells. In addition to IFN-γ, cNK cells release perforin and granzymes to induce apoptosis of target cells. ILC2s are defined as ILCs that express GATA-binding protein 3 and produce such cytokines as IL-4, IL-5, IL-9 and IL-13 as well as the epidermal growth factor, amphiregulin; like Th2 cells, they are considered to be involved in anti-helminth immunity. ILC3s are defined as ILCs that express retinoic acid receptor-related orphan receptor-γt and produce cytokines such as IL-17A, IL-22 and granulocyte macrophage colony-stimulating factor (GM-CSF); they include both natural cytotoxicity receptor (NCR)− ILC3s and NCR+ ILC3s, and are considered to be involved in anti-bacterial immunity, like Th17 cells.
Recently, another ILC subset called regulatory ILCs (ILCregs), which resemble regulatory T cells (Tregs) and have regulatory functions, has been reported.123 ILCregs produce regulatory cytokines such as IL-10 and/or transforming growth factor-β (TGF-β), but they do not express Forkhead box protein P3 which is the canonical transcription factor of Tregs. Therefore, it remains controversial whether ILCregs represent an independent effector subset, like ILC1s, ILC2s and ILC3s, or just a temporary state of ILCs.
ILCs are generally thought to be tissue-resident cells that differentiate into mature effector cells in tissues and show minimal movement between organs.4 Instead, they have functional plasticity that enables them to respond promptly to microenvironmental changes, thereby precluding any need for differentiation and/or migration of new ILC subsets adapted to a new environment. For instance, transdifferentiation has already been shown between ILC1s and ILC3s,56 between ILC1s and ILC2s,789 and between ILC2s and ILC3s (Fig. 1).1011
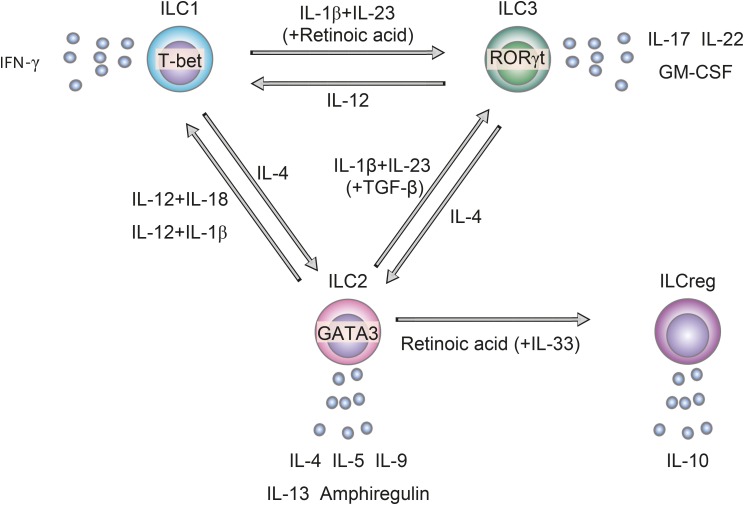 | Fig. 1Plasticity of ILCs in the airways. IL-4 induces conversion of ILC1s and ILC3s to ILC2s. IL-12, in conjunction with IL-1β or IL-18, induces conversion of ILC2s to ILC1s. IL-1β and IL-23, in conjunction with TGF-β, induce conversion of ILC2s to ILC3s. IL-1β and IL-23 also induce conversion of ILC1s to ILC3s in the intestines, but this has not been demonstrated in the airways. Retinoic acid, in conjunction with IL-33, induces conversion of ILC2s to ILCregs.ILC, innate lymphoid cell; ILC1, group 1 innate lymphoid cell; ILC2, group 2 innate lymphoid cell; ILC3, group 3 innate lymphoid cell; IL, interleukin; TGF-β, transforming growth factor-β; ILCreg, regulatory innate lymphoid cell; IFN, interferon; T-bet, T-box-expressed-in-T cells; RORγt, express retinoic acid receptor-related orphan receptor-γt; GATA3, GATA binding protein 3.
|
Go to :
DISTRIBUTION, HETEROGENEITY AND TISSUE-SPECIFIC SIGNATURES OF ILCs
ILCs are present in various organs throughout the body, and the proportions of the subsets differ depending on the organ. In humans, ILC3s are the predominant population in mucosal tissues, including the lung and gut,7 whereas the proportion of ILC2s is a little higher in the skin compared to mucosal tissues.7 The proportions of the ILC subsets are also influenced by age. For instance, although ILC3s (including NCR− ILC3s and NCR+ ILC3s) are the predominant population in the fetal human lung, their proportion decreases while the proportions of ILC1s and ILC2s increase with age in the adult human lung.7 Another important point is that there is substantial heterogeneity in each subset of ILCs. Moreover, ILCs show different phenotypes depending on the organ.12 For instance, although ILC2s from different organs share canonical markers such as GATA3 and IL-7R, their expressions of IL-33R, IL-25R and IL-18R1 differ depending on the organ.12 Such heterogeneity is probably due to the high plasticity of ILCs, which enables them to adjust to their microenvironment.
Go to :
CYTOKINES AND OTHER FACTORS ASSOCIATED WITH EACH ILC SUBSET IN THE AIRWAYS ILC1s
IL-12
IL-12 is known as a major activator of ILC1s and promotes their secretion of IFN-γ.6 The major physiological producers of IL-12 are antigen-presenting cells (APCs) such as dendritic cells (DCs) and macrophages. In the mouse lung, IFN-γ produced by ILC1s in response to DC-derived IL-12 during viral infection suppresses early viral growth,13 suggesting that the IL-12–ILC1 axis may be involved in anti-viral immunity (Fig. 2). Furthermore, IL-12 mediates the transdifferentiation of ILC2s789 and ILC3s5 into IFN-γ-producing ILC1s (Fig. 1), a mechanism that may be involved in immune responses to viral infections and in the pathophysiology of chronic obstructive pulmonary disease (COPD).
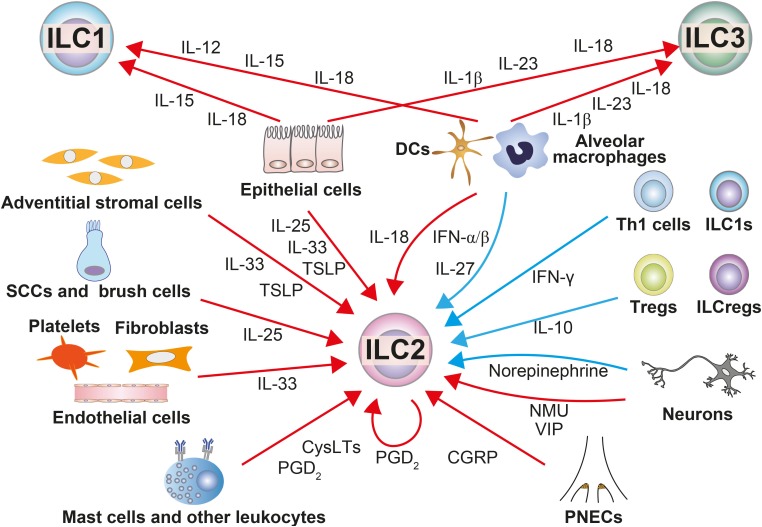 | Fig. 2Regulators of ILCs in the airways and their sources. In the airways, ILCs are regulated by various stimuli that are produced by other cells. The red arrows indicate stimuli that activate ILCs, and the blue ones indicate stimuli that suppress ILCs.ILC, innate lymphoid cell; ILC1, group 1 innate lymphoid cell; ILC2, group 2 innate lymphoid cell; ILC3, group 3 innate lymphoid cell; IL, interleukin; ILCreg, regulatory innate lymphoid cell; IFN, interferon; DC, dendritic cell; Th, T helper; Treg, regulatory T cell; SCC, solitary chemosensory cell; TSLP, thymic stromal lymphopoietin; CysLT, cysteinyl leukotriene; PGD2, prostaglandin D2; NMU, neuromedin U; VIP, vasoactive intestinal peptide; CGRP, calcitonin gene-related peptide; PNEC, pulmonary neuroendocrine cell.
|
IL-15
Like IL-12, IL-15 activates ILC1s to produce IFN-γ. IL-15 is known to be produced by APCs, a subset of thymic epithelial cells, and by stromal cells. In the airways, human bronchial epithelial cells produce IL-15 in response to respiratory syncytial virus infection (Fig. 2).14 In human airway diseases, IL-15–positive cells have been reported to be increased inpatients with sarcoidosis, tuberculosis or chronic bronchitis compared to asthmatic patients and healthy subjects,15 suggesting the involvement of IL-15 in the pathophysiology of these diseases.
IL-18
IL-18 was once thought to be just an activator of ILC1s that induces IFN-γ production. However, recent studies have shown that IL-18 also activates ILC2s and ILC3s to produce their signature cytokines,1216 suggesting that IL-18 may be a pan-activator of ILCs (Fig. 2). Furthermore, IL-18 and IL-12 together promote conversion of ILC2s to ILC1s (Fig. 1).8 IL-18 is produced by APCs such as macrophages and DCs. In regard to the airways, IL-18 was shown to be released from human bronchial epithelial cells upon human rhinovirus infection17 and Alternaria extract stimulation18
in vitro. In addition, cigarette smoke exposure induced IL-18 production by alveolar macrophages in the mouse lungs (Fig. 2).19 In humans, the levels of IL-18 in bronchoalveolar lavage fluids (BALFs) were significantly higher in patients with COPD than in healthy subjects, and even higher in patients with acute exacerbation of COPD.20 In addition, the expression of IL-18 in lung epithelial cells was significantly increased in patients with severe COPD compared to healthy individuals who never smoked.17 These findings suggest that IL-18 may be involved in the pathophysiology of COPD.
ILC2s
IL-25
IL-25 activates ILC2s and promotes type 2 cytokine production.21 Various kinds of immune cells, such as macrophages, eosinophils and T cells, have been shown to produce IL-25. Recently, bottle-shaped epithelial-lineage cells expressing taste receptors, named tuft cells—including intestinal tuft cells, brush cells in the lower airways and solitary chemosensory cells (SCCs) in nasopharyngeal tissue—have attracted broad attention as major sources of IL-25.21 In mice, intestinal tuft cells produce IL-25 after sensing microbial metabolites through succinate receptors or taste receptors during protozoan and helminth infections, which results in activation of ILC2s and promotion of an anti-helminth response.21 Similarly, recent findings have suggested that SCCs in the human upper respiratory tract22 and brush cells in the murine lower respiratory tract23 are major producers of IL-25 in the airways (Fig. 2). In humans, IL-25 messenger RNA (mRNA) expression in bronchial epithelial cells was significantly increased in a subgroup of asthmatic patients with a Th2-high phenotype.24 In addition, the levels of IL-25 were significantly increased in nasal mucosal fluid from asthmatic patients during RV infection compared to healthy individuals.25 In mice, IL-25–producing brush cells were increased in the lungs of mice challenged with leukotriene (LT) E4, Alternaria or house dust mites, resulting in asthma-like airway inflammation.23 Moreover, epithelial cell-derived IL-25 has been shown to be crucial in an ovalubumin (OVA)-induced asthma model,26 suggesting that IL-25 may play a critical role in the development of asthma. IL-25-producing SCCs22 and ILC2s272829 were increased in nasal polyps from chronic rhinosinusitis with nasal polyps (CRSwNP) compared to nasal tissue from healthy individuals, and the level of IL-25 correlated with infiltration of inflammatory cells and expression of inflammatory markers.30 These findings suggest that the IL-25–ILC2 axis may be involved in the pathophysiology of CRSwNP. Besides allergic disorders, the concentration of IL-25 and the number of ILC2s were increased in BALF from patients with idiopathic pulmonary fibrosis and in the lungs of mice with helminth–induced lung fibrosis compared to controls,31 suggesting possible involvement of the IL-25–ILC2 axis in lung fibrosis as well.
IL-33
Unlike other cytokines that are newly synthesized upon stimulation and secreted via the endoplasmic reticulum/Golgi pathway, IL-33 is constitutively expressed in cells at the mucosal barrier and released from the nucleus in active form in response to tissue damage.32 Therefore, it is believed to be one of the “alarmins” that gather components of the repair response to the sites of injury. However, several studies suggest that IL-33 may be actively secreted from live cells, including bronchial epithelial cells33 and fibroblasts, even in the absence of necrosis (Fig. 2). Although the mechanisms of IL-33 secretion are not fully understood, adenosine triphosphate-induced purinoceptor-dependent activation of epithelial nicotinamide adenine dinucleotide phosphate oxidase, i.e., dual oxidase 1, may be involved.33 IL-33 is recognized as one of the major activators of ILC2s that induce production of type 2 cytokines. In mice, IL-33 is released from alveolar epithelial cells in response to tissue damage32 caused by fungi such as Alternaria and Aspergillus and viruses such as RSV and RV.32 Meanwhile, in humans, IL-33 is released from bronchial epithelial cells located more centrally,32 similar to IL-25 and thymic stromal lymphopoietin (TSLP) (Fig. 2).
The expression of IL-33 in the lungs peaks during infancy, and declines with age. In line with that, the number of ILC2s in the lungs also peaks in infancy.34 These findings suggest that IL-33 may play a major role in the developing phase of acquired immunity and that epithelial damage may induce more severe allergic airway inflammation during infancy than during adulthood through the IL-33–ILC2s axis. Furthermore, the expression of IL-33 in the nucleus of airway epithelial cells is enhanced by exposure to atmospheric substances such as cigarette smoke32 and diesel exhaust particles.35 This suggests that damage to epithelial cells after exposure to cigarette smoke and diesel exhaust particles evokes release of larger amounts of IL-33, which results in aggravation of allergic inflammation. In addition to epithelial cells, stromal cells,36 endothelial cells, fibroblasts32 and platelets37 may produce IL-33 (Fig. 2). In agreement with the finding that the IL-33 gene was strongly associated with asthma susceptibility in several genome-wide association studies,38 the level of IL-33 was higher in BALF from asthmatic patients than in controls and showed an inverse correlation with forced expiratory volume in one second (FEV1), regardless of the history of steroid treatment.39 The levels of IL-33 in serum40 as well as in exhaled breath condensate were higher in COPD patients than in healthy individuals and correlated with the eosinophil counts in the peripheral blood.41 Although it remains unclear whether IL-33 is involved in the pathogenesis of COPD, these findings suggest such involvement, especially in patients with eosinophilic COPD.
TSLP
Like other epithelial–derived cytokines such as IL-33 and IL-25, TSLP is recognized as a major activator of ILC2s that induces production of type 2 cytokines. However, unlike other epithelial-derived cytokines, TSLP was shown to induce corticosteroid resistance in murine ILC2s through activation of an intracellular signaling molecule, signal transducer and activator of transcription 5.42 TSLP is produced by various kinds of cells including DCs, vascular endothelial cells, macrophages and mast cells. In the airways, similar to IL-25 and IL-33, TSLP is produced mainly by airway epithelial cells in response to exposure to bacteria, fungi and viruses.43 Adventitial stromal cells localize with ILC2s in adventitial niche around the lung bronchi and large vessels, and support ILC2s through constitutive expression of TSLP and IL-33.36 In humans, the number of type 2 cytokine-producing ILC2s was significantly increased in sputum from patients with corticosteroid-resistant severe asthma compared those with mild asthma, whereas the number of type 2 cytokine-producing Th2 cells was comparable in both groups. The levels of TSLP in BALF from patients with asthma correlate inversely with steroid-mediated inhibition of IL-5-producing ILC2s in the BALF. Furthermore, a clinical trial of an anti-TSLP antibody, tezepelumab, showed that, in moderate to severe steroid-resistant asthma, anti-TSLP treatment significantly reduced asthma exacerbation as well as important biologic markers such as the blood eosinophil count and fractional exhaled nitric oxide (FeNO).44 These findings suggest that the TSLP–ILC2 axis may play critical roles in steroid resistance in asthmatic patients and that TSLP is likely to be continuously produced and released in the airways of moderate to severe asthmatic patients.
IL-27
IL-27 is generally produced by DCs and macrophages (Fig. 2). In mice, IL-27 suppresses the proliferation and cytokine production of ILC2 cells in vitro,445 and it also suppresses Alternaria-induced eosinophilic airway inflammation by regulating ILC2 activation in vivo.4 However, in humans, IL-27 mRNA levels and the percentage of IL-27-expressing cells in BALF were increased in patients with asthma, especially those with a severe phenotype.46 Therefore, the involvement of IL-27-ILC2s in the pathophysiology of asthma in humans remains to be elucidated.
IFNs
IFNs are divided into types 1 (α/β), 2 (γ) and 3 (λ). Type 1 and 2 IFNs have been shown to suppress type 2 cytokine production by ILC2s, both in vitro and in vivo.445 The major producers of IFN-α and -β are macrophages and DCs. IFN-γ is produced by activated Th1 cells and ILC1s, including NK cells, which are activated mainly through TLRs (Fig. 2). In mice, the deficiency of type 1 IFN during influenza virus and helminth infections results in severe or prolonged eosinophilic airway inflammation mediated by activated ILC2s. In humans, dozens of reports have shown impaired production of type 1 and 3 IFNs by cultured primary bronchial epithelial cells, BAL cells, peripheral blood mononuclear cells (PBMCs) and plasmacytoid DCs in response to infection with viruses such as RSV and RV in patients with asthma compared to healthy individuals.47 Therefore, dysregulation of ILC2 activity by type 1 and 3 IFNs during viral infection in asthmatic patients may result in the development and exacerbation of allergic airway inflammation.
Lipid inflammatory mediators
Lipids are primarily involved in the formation of cell membranes of organs. However, some reports have shown that certain lipids also play crucial roles in immune responses and the maintenance of homeostasis. These lipids are called bioactive lipids or lipid mediators. Cysteinyl leukotrienes (CysLTs) as well as prostaglandin (PG) D2 are products of arachidonic acid and were known to be major pro-inflammatory lipid mediators of allergic disorders from early days. Mast cells activated by immunoglobulin (Ig) E-crosslinking are the major source of PGD2 in terms of quantity, but other leukocytes, including eosinophils, Th2 cells, DCs and cytokine-activated ILC2s,48 also produce PGD2 (Fig. 2). Since human ILC2s are identified as lineage-negative cells expressing chemoattractant receptor-homologous molecules on Th2 cells (CRTH2),29 which is the PGD2 receptor, PGD2 influences ILC2s in a variety of ways, including their migration49 and production of IL-13.50 CysLTs are generally produced by leukocytes such as eosinophils, mast cells, macrophages and basophils. CysLTs act directly on ILC2s to enhance their ability to produce type 2 cytokines, both in vivo and in vitro.50 On the other hand, there are some lipid molecules that inhibit ILC2 activation. PGI2, PGE2 and lipoxin A4—also products of arachidonic acid—suppress ILC2s' cytokine production and proliferation, in vitro and in vivo.50 Interestingly, however, LTE4 and PGD2 reportedly induce Th2 cytokines, including IL-4, synergistically in purified human peripheral blood ILC2s.51
Neuropeptides
Neuropeptides are peptides that are expressed in the nervous system and exhibit physiological activity. They are present not only in the central nervous system, but also in the nervous system of peripheral tissues such as the lungs, and they function as signal transmitters between cells. Among several neuropeptides known to act on ILC2s, vasoactive intestinal peptide (VIP) was the first one shown to modulate ILC2 activation. VIP belongs to the glucagon/secretin family and is highly expressed in intestinal neurons, where it coordinates pancreatic secretion with smooth muscle relaxation in response to feeding. Both lung and intestinal ILC2s express VIP receptors, including VIP receptor type 1 and type 2, and VIP simulation induces IL-5 production by the cells. The IL-5 produced in turn activates sensory neurons to produce VIP,52 which may exacerbate allergic airway inflammation (Fig. 2). Lung ILC2s also express receptors for another neuropeptide, called neuromedin U (NMU), whereas ILC1s and ILC3s do not. NMU is thought to directly activate lung ILC2s to proliferate and produce type 2 cytokines.53 Calcitonin gene-related peptide (CGRP) is a calcitonin gene product, like the thyroid hormone calcitonin and it is involved in the regulation of blood calcium levels. CGRP is widely distributed in the central and peripheral nervous systems; It was also produced by non-neuronal cells in the airways—called pulmonary neuroendocrine cells (PNECs)─after OVA challenge in an OVA-sensitized mouse model (Fig. 2).54 Interestingly, a recent study has shown that ILC2s are localized in close proximity to PNECs and that CGRP enhances type 2 cytokine production by lung ILC2s in the presence of IL-33 or IL-25,54 suggesting that interaction between PNECs and ILC2s may be involved in allergic airway inflammation. Besides the neuropeptides that induce activation of ILC2s, there is also a neuropeptide that regulates activation of ILC2s. Both lung and intestinal ILC2s express the β2-adrenergic receptor (β2-AR), which is a receptor for epinephrine released by sympathetic nerve stimulation. Treatment with a β2-AR agonist, salmeterol, suppressed proliferation and type 2 cytokine production by ILC2s in an IL-33-induced airway inflammation model (Fig. 2).55 These findings suggest that β2-AR agonists used as therapeutic agents for asthma may work not only as a bronchodilator, but also as a suppressor of type 2 inflammation induced by ILC2s.
Sex steroids
Sex steroids, such as estrogen and androgen, are steroid hormones that are produced mainly by the reproductive organs and modulate reproductive functions. In addition to their effects on the reproductive organs, sex steroids have recently been shown to have effects on immune cells, including ILC2s in peripheral tissues. Androgen receptors are expressed on lung ILC2s56 as well as ILC2 progenitors (ILC2Ps) in bone marrow (BM),57 whereas estrogen receptors are expressed on lung ILC2s and uterine ILC2s,58 but not on ILC2Ps in BM.57 These findings indicate that androgens may influence both the development of ILC2s in BM and the activation of ILC2s in peripheral tissues, whereas estrogens may influence mainly ILC2s in peripheral tissues. Androgens and estrogens are thought to exert opposite effects on ILC2s. Androgen signaling inhibits differentiation of ILC2Ps into ILC2s57 and also activation of ILC2s.5657 In contrast, estrogen has been suggested to have supportive effects on ILC2s (Fig. 2).58 Indeed, the numbers of lung ILC2s57 and BM ILC2Ps are significantly lower in adult male mice than in adult female mice in the steady state.57 In addition, the number of ILC2s in the peripheral blood of adult male patients with severe asthma,56 as well as their number in the lungs of Alternaria-exposed adult male mice,56 was significantly lower than in females. In humans, although males are more susceptible to asthma than females before puberty, the asthma incidence reverses after puberty.59 These findings suggest that ILC2s are involved in the sex difference in asthma prevalence in humans.
ILC3s
IL-23
IL-23 is a major activator of ILC3s that induces production of inflammatory cytokines such as IL-17 and IL-22. IL-23 also induces conversion of ILC1s to ILC3s in conjunction with IL-1β and retinoic acid,5 and ILC2s to ILC3s in conjunction with IL-1β and TGF-β (Fig. 1).1011 IL-23 is generally produced by DCs and macrophages, but it was also produced by bronchial epithelial cells in a house dust mite-induced asthma model (Fig. 2).60 The serum level of IL-23 was higher in asthmatic children than in healthy children, which shows a strong inverse correlation with FEV1.61 In addition, IL-23-positive cells were increased in the airway mucosa of stable COPD patients.62 However, the roles of IL-23 in the pathogenesis of asthma and COPD remain unclear.
IL-1β
IL-1β is a major activator of ILC3s that induces IL-17A production.63 While IL-1β is a potent activator of ILC2s that induce type 2 cytokine production,7 it also induces conversion of ILC2s to ILC1Ss together with IL-12,79 and to ILC3s together with IL-23 and TGF-β (Fig. 1).11 In the airways, IL-1β is produced by DCs in response to exposure to chitin and IL-33,64 and by nasal epithelial cells exposed to Staphylococcus aureus or Pseudomonas aeruginosa (Fig. 2).11 The levels of IL-1β in BALF and expression of mRNA for IL-1β and NLRP3 in sputum cells65 were significantly higher in steroid-resistant severe asthma patients with neutrophilic inflammation than in healthy subjects and mild asthmatic patients. In mice, steroid-resistant severe asthma-like airway inflammation was attenuated by inhibiting IL-1β or inflammasome activation.65 These findings suggest that IL-1β is involved in the pathogenesis of steroid-resistant severe asthma. In contrast, the levels of IL-1β in BALF and IL-1β mRNA expression in the bronchial mucosa were comparable between severe COPD patients and control subjects.66 In addition, a clinical trial of anti-IL-1R1 monoclonal antibody (mAb) showed no improvement in the QOL score or acute exacerbation rate in COPD patients.67 Therefore, the question of involvement of IL-1β in the pathogenesis of COPD should be further studied.
Vitamins
Retinoic acid (RA)–which is a metabolite of vitamin A (Vit A)–and vitamin D (Vit D) are known to be molecules that regulate ILCs.1351168 RA is synthesized from a Vit A metabolite, retinal, by cells having enzymes such as retinaldehyde dehydrogenase (ALDH)1A1, ALDH1A2 and ALDH1A3. RA is generally synthesized by CD103+ DCs, intestinal epithelial cells and lamina propria stromal cells in the gut that express ALDHs. In the airways, bronchial epithelial cells express ALDHs in response to IL-13 stimulation,1 suggesting that these cells could be the source of RA during allergic airway inflammation. Vit D can be absorbed by oral intake, but it is synthesized mainly in the skin upon exposure to ultraviolet light from the sun. RA enhances activation of ILC3s by IL-1β and IL-23 to increase production of IL-22 (Fig. 2), and it also induces conversion of ILC1s to ILC3s in conjunction with IL-1β and IL-23 (Fig. 1).5 In addition, RA inhibits development of ILC2s from ILC2Ps in mouse BM69 and induces conversion of ILC2s to IL-10–producing ILCregs in both humans and mice (Fig. 1).13 In contrast to the positive effects of RA on ILC3s, Vit D suppresses production of cytokines such as IL-22, IL-17F and GM-CSF by ILC3s by down-regulating the IL-23/IL-23R pathway,68 and it also prevents IL-1β-, IL-23- and TGF-β-induced conversion of ILC2s to ILC3s (Fig. 1).11
Involvement of ILCs in airway inflammatory diseases
Asthma
Asthma is a chronic airway inflammatory disease characterized by wheezing, prolonged exhalation and dyspnea due to reversible airflow obstruction. It is considered to be a heterogeneous disease comprising a number of subtypes.70 ILC2s might be involved in the pathogenesis of asthma in patients who have severe eosinophilic inflammation induced by type 2 cytokines. Indeed, the frequencies of ILC2s in BALFs, sputum and PBMCs were significantly increased in asthmatic patients, especially in uncontrolled patients, compared to healthy individuals, and correlated negatively with airway function.71 ILC2s may be involved in the pathogenesis of asthma via 2 different mechanisms: antigen non-specific and antigen specific. The former involves direct activation of ILC2s by mediators such as IL-33, TSLP, IL-25 and neuropeptides that are produced by tissue structural cells in response to microenvironmental changes such as viral infection, allergen exposure and stress. The latter involves migration and activation of ILC2s by lipid mediators such as PGD2 and CysLTs, which are synthesized by mast cells activated upon antigen exposure in an IgE-dependent manner. The number of ILC2s in BALFs of asthmatic patients is increased after segmental allergen challenge, whereas the number of ILC2s in the blood is decreased.49 Moreover, the levels of PGD2 in BALFs correlated with decreased ILC2s in the blood.49 Although ILCs are recognized as tissue-resident cells that show minimal migration between organs in mice, these findings suggest that, at least in humans, some ILC2s dynamically migrate into the airways from the peripheral blood in response to PGD2 that is synthesized by mast cells upon allergen exposure.
As the importance of ILC2s and its activators in the pathogenesis of asthma becomes apparent, new therapeutic strategies—including the use of biologics to suppress their activity—are attracting attention. Treatment with an anti-TSLP mAb, tezepelumab, was shown to reduce bronchoconstriction and airway inflammation induced by allergen challenge in mild asthmatic patients.72 That mAb also significantly reduced the annualized rate of asthma exacerbations in steroid-resistant severe asthmatic patients treated with long-acting β-agonists and medium-to-high doses of inhaled glucocorticoids in a phase 2 clinical trial.44 Of note, biomarkers such as the blood eosinophil count and FeNO were significantly decreased as early as 4 weeks after treatment with tezepelumab,44 suggesting that TSLP is continuously synthesized and released in the airways of asthmatic patients. Since TSLP is a key molecule inducing steroid resistance of ILC2s42 as described in the previous section, treatment with tezepelumab may be the most logical strategy to treat steroid-resistant severe asthmatic patients. Another biologic directed at an epithelial cell-derived cytokine, anti-IL-33 antibody, is currently undergoing a phase 2a clinical trial in asthmatic patients being treated with high-dose inhaled corticosteroid and long-acting β2-agonist. Fevipiprant, a CRTH2 antagonist that targets CRTH2 on ILC2s as well as on eosinophils, basophils and Th2 cells, significantly reduced eosinophilic inflammation in the sputum and bronchial mucosa and improved the lung function and clinical outcome in week 12 in moderate to severe eosinophilic asthmatic patients.73 However, 2 studies using different CRTH2 antagonists, AZD 198174 and OC000459,75 showed no significant improvement in lung function in moderate, persistent asthma patients in week 4. Therefore, further studies are needed to determine the patient phenotypes likely to benefit from use of a CRTH2 agonist, and the duration of treatment. In contrast to the involvement of ILC2s in eosinophilic asthma, ILC3s may be involved in non-allergic asthma, including obesity-related asthma. In mice, airway hyperresponsiveness (AHR) was induced in high-fat-diet-fed obese mice even in the absence of antigen challenge compared to normal-diet-fed mice.63 AHR was induced by IL-17-producing ILC3s activated by IL-1β produced upon obesity-related inflammasome activation.63 However, in humans, the details of any involvement of ILC3s in non-eosinophilic asthma remain unclear.
Allergic rhinitis (AR)
AR is an allergic disorder of the nasal mucosa that is characterized by paroxysmal recurrent sneezing, serous rhinorrhea and nasal congestion. AR is thought to be induced mainly by an immediate hypersensitivity reaction that involves IgE-mediated release of histamine and other mediators from mast cells in the nose. However, a delayed allergic reaction accompanied by production of type 2 cytokines that is triggered by inhalation of a sensitizing antigen may also be involved in the development of the symptoms and pathology of AR. It is thus assumed that ILC2s may also be involved via type 2 cytokine production. Indeed, the proportion of ILC2s in the peripheral blood of patients with seasonal AR is significantly higher in the pollen season than out of season.76 In addition, ILC2s sorted from the peripheral blood of AR patients produce larger amounts of type 2 cytokines than those from healthy individuals.77 These findings suggest that ILC2s in patients with AR may be activated upon allergen exposure. Like in asthma, ILC2s may be involved in the pathogenesis of AR via 2 different mechanisms: antigen non-specific and specific. The former involves direct activation of ILC2s by mediators such as TSLP and IL-25, which are increased in the nasal lavage of patients with AR. The latter involves migration and activation of ILC2s in response to lipid mediators such as PGD2 and CysLTs, synthesized by mast cells activated in an IgE-dependent manner upon antigen exposure. The proportion of ILC2s in the peripheral blood of patients who underwent allergen-specific immunotherapy did not increase even during the pollen season.76 That may be due to desensitization of mast cells by the immunotherapy, resulting in decreased release of PGD2 and CysLTs by the mast cells.
Chronic rhinosinusitis (CRS)
CRS is a heterogeneous disease characterized by chronic inflammation of the sinus and nasal mucosa, with nasal congestion and a decreased sense of smell. CRS can be roughly divided into 2 major subtypes based on the presence/absence of nasal polyps, and each subset shows different pathological conditions.78 In particular, CRSwNP is generally considered to involve mainly eosinophilic inflammation caused by typical type 2 cytokines, with high expression of IL-5 and IL-13 in polyps. The percentage of ILC2s is significantly higher in nasal polyps from patients with CRSwNP than in nasal tissues from controls and those with chronic rhinosinusitis without nasal polyps (CRSsNP).272829 The percentage correlates with the nasal symptom score.27 Furthermore, unlike ILC2s in the peripheral blood, ILC2s sorted from nasal polyps of CRSwNP spontaneously produce type 2 cytokines such as IL-5 and IL-13, even without additional stimulation.28 These findings indicate that ILC2s in CRSwNP patients are continuously exposed to activating signals in the nasal cavity.
Although factors that continuously activate ILC2s in nasal polyps remain unclear, recent findings highlighted a substantial role of IL-25 in CRSwNP, especially in polyps having high eosinophil infiltration.2230 The expression of IL-2530 and the number of IL-25-producing SCCs22 were significantly higher in nasal polyps from CRSwNP patients than in nasal tissue from controls and CRSsNP patients. In addition, production of IL-25 by SCCs in nasal polyps was increased in response to stimulation with IL-13,22 suggesting that a vicious loop of IL-25-ILC2s-IL-13 may be present. Recent studies have shown that the pathophysiology of CRSwNP may differ depending on the race, implying the existence of subtypes in CRSwNP.78 Indeed, it has been shown that CRSwNP patients can be divided into 2 subtypes depending on the expression level of IL-25 in nasal tissues. The IL-25-high subtype had a type 2-prone phenotype accompanied by high eosinophil infiltration and steroid sensitivity.79 TSLP was also constantly up-regulated in nasal tissue from CRSwNP patients compared to healthy subjects.80 In contrast, there is still debate regarding the expression level of IL-33 in nasal polyps.8081 In regard to treatment, a recent case report demonstrated that an IL-4R mAb (dupilumab) significantly improved CRSwNP disease outcomes, along with asthma control and lung function, in patients with CRSwNP and aspirin-exacerbated respiratory disease.82 A clinical phase 2 trial of ANB020, an anti-IL-33 antibody, is currently ongoing.
COPD
COPD is a chronic airway inflammatory disease characterized by dyspnea due to irreversible airway obstruction. Cigarette smoking is considered to be the primary cause of COPD, but continuous inhalation of air pollutants and occupational dust may also be involved. COPD is thought to be a heterogeneous disease that includes patients with chronic bronchitis, emphysema and/or asthma components. Although the pathogenesis of COPD may also be heterogeneous, type 1 and 3 immunity accompanied by neutrophil and macrophage infiltration is thought to be involved.83 Indeed, the frequency of ILC1s in the peripheral blood is increased in COPD patients and correlates inversely with lung function.8 In addition, the frequencies of ILC1s and NCR− ILC3s were increased in lung tissue of COPD patients compared to control subjects, while the frequency of ILC2s was decreased.7 Given that the total ILC numbers are comparable between healthy individuals and COPD patients, the increase in ILC1 frequency and decrease in ILC2 frequency have been proposed to be caused by conversion of ILC2s to ILC1s in the lung rather than influx of ILCs from other organs.7 With regard to ILC3s, a subset of ILC3s that express neuropilin 1 was increased in the lung tissue of COPD patients, and these cells were suggested to be involved in ectopic lymphocyte accumulation and angiogenesis.84 In line with these findings in humans, the numbers of ILC1s and ILC3s were reportedly increased in lungs from a cigarette smoke-induced COPD mouse model.85 These findings suggest that ILC1s and ILC3s, but not ILC2s, are involved in the pathophysiology of COPD. However, as mentioned above, COPD is thought to be a heterogeneous disease, and accumulating evidence suggests the existence of subtypes involving type 2 immunity. For instance, it has been shown that some COPD patients have eosinophilic inflammation during acute exacerbation.86 Furthermore, some patients have features of both asthma and COPD, called asthma-COPD overlap, with increased FeNO and eosinophils in the peripheral blood.87 Although type 2 immunity may be involved in the pathophysiology of these patients, any possible involvement of ILC2s in it remains unclear.
Go to :
CONCLUSIONS
Many articles regarding the functions and regulators of ILC2s have been published in recent years, resulting in those features being relatively well characterized. On the other hand, articles regarding ILC1s and ILC3s have been scanty in number for characterizing their functions and regulators. One of the reasons why analysis of ILC2s is advanced compared to the other ILC subsets may be that ILC2 activation leads to a clear outcome: eosinophil infiltration of tissues. Indeed, many of the studies showing involvement of ILC2s consist of patients with severe eosinophil infiltration. For such patients, treatments that target such upstream regulators of ILC2s as TSLP, IL-25 and IL-33, and downstream effector molecules of ILC2s, such as IL-4, IL-5 and IL-13, can be thought to have promise. However, in clinical practice, there are many cases that involve not only pure eosinophil infiltration but also neutrophil infiltration, and such “mixed-type” patients often exhibit more severe and treatment-resistant phenotypes. Since not only ILC2s but also ILC1s and ILC3s may be involved in the pathophysiology of such cases, we need to further elucidate functions and molecules that modulate activation of ILC1s and ILC3s in the airways. Recently, IL-17-producing ILC2s expressing the canonical ILC2 surface marker ST2 were reportedly induced in the lungs of papain- or IL-33-treated mice,88 suggesting the existence of cells intermediate between 2 different ILC subsets in vivo. Conventional analysis using canonical surface markers and cytokines would not be enough to clarify the characteristics and functions of ILCs involved in the pathophysiology of “mixed-type” airway inflammatory disease. Analysis of airway ILCs at the single-cell levels using a recently developed technology, single cell RNA-seq may help determine the ILC phenotype and local microenvironment in individual cases.
Go to :
 XML Download
XML Download