

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Primary hepatic neuroendocrine tumours (PHNETs) were described by Edmondson in 1958.1 They are rare primary hepatic tumours that constitute 0.3% of all neuroendocrine tumours, with fewer than 150 cases reported in literature till date.23 PHNET is a diagnosis of exclusion and evaluation continues over pre-operative, intra-operative and post-operative period, being a diagnosis in continuum.3 The picture is further complicated by non-specific clinical features and overlapping imaging features with other common primary and metastatic liver cancers.2 Although clinical features and imaging characteristics have been explained in detail in various case series, information is scarce with respect to treatment options and survival outcomes.123 We have reviewed the clinical data of patients with PHNETs to study the clinical presentations, imaging features, treatment options and survival outcomes.
Go to : 
MATERIALS AND METHODS
A retrospective analysis of prospectively maintained liver cancer database from January 2009 to August 2018 was performed at Tata Memorial Centre, Mumbai. Electronic medical records of patients with diagnosis of PHNETs were studied for clinical features, imaging characteristics, type of surgical resection, post-surgical treatment, non-surgical treatments, overall survival (OS) and disease free survival (DFS) outcomes. Institutional review board clearance was taken for review of electronic medical records (IEC/1217/2003/001). In order to establish the diagnosis of PHNETs, an extensive investigational protocol was followed, that included history and physical examination, cross-sectional imaging, full body fusion metabolic scans, bidirectional endoscopies, endoscopic ultrasonography and in surgical group in addition to above mentioned investigations, intra-operative ultrasound of pancreas and duodenum along-with intra-operative organ palpation particularly small bowel to rule out any latent primary or extra hepatic disease. All patients were initially evaluated with a dynamic computed tomography of chest, abdomen and pelvis, supplemented by gallium-68-labeled [1, 4, 7, 10-tetraazacyclododecane-1, 4, 7, 10-tetraacetica cid]-1-Nal3-octreotide (68Ga-DOTA-NOC) positron emission tomography (PET) scan to rule out extra-hepatic disease or detect any latent primary. Bidirectional endoscopies and endoscopic ultrasonography were done as a part of standard protocol to rule out any extra hepatic primary. All patients were discussed in multidisciplinary joint clinic meetings and treatment strategy was formulated based on tumour grade and burden that was determined by the extent of liver involvement, size of the lesions and number of the lesions. Follow up was updated until October 2018. Follow up protocol was standardised and included 3 monthly serum chromogranin levels with abdominopelvic ultrasonography and 6 monthly 68Ga-DOTA-NOC PET scans to detect any latent primary or recurrence. Survival analysis was done using Statistical Package for the Social Sciences. (Version 20.0, SPSS) For the purpose of survival analysis patients were divided into surgical and non-surgical treatment groups. Overall and disease free survival was compared between tumour grades and Ki 67 indices by using log rank test.
Go to :
RESULTS
Demographics and clinical features
PHNETs constituted 0.6% of all the liver tumours in our database, with a median age of 50 years (range 14–65 years) and a male: female ratio of 9:4. In our series, the clinical presentation was abdominal pain in 5 (38%) patients, incidental in 5 (38%) and 3 (24%) patients had diarrhoea and breathlessness as the predominant symptom. One of the patients had underlying primary biliary cirrhosis and one was previously treated for cervical cancer (Table 1).

Tumour markers and imaging findings
All patients had normal Carcinoembryonic Antigen (CEA) and Alpha-fetoprotein (AFP) levels; while Carbohydrate antigen (CA 19-9) was marginally raised in three patients with a median of 71 IU/L (range 64–86). Median Chromogranin A levels were 700 ng/L (range 100–10933 ng/L). Dynamic computed tomography imaging findings were varied with most predominant pattern being hypo dense multi-cystic mass with rim enhancement in 6 (47%) patients followed by solid hypo-enhancing features in 5 (38%) patients and solid arterially enhancing lesions with delayed washout in 2 (15%) patients. DOTANOC activity was absent in 1 patient with Grade III NEC (neuroendocrine carcinoma), however FDG activity was noted in the same. None of the patients had extra-hepatic disease on 68Ga-DOTA-NOCor FDG PET scan (Fig. 1).
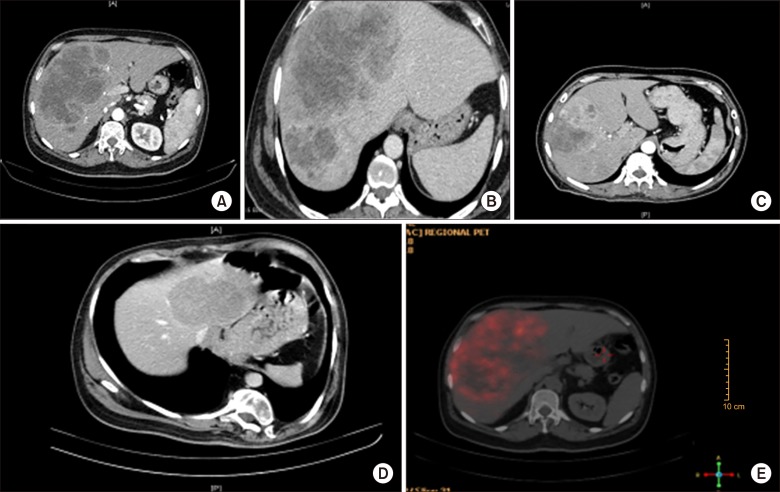
Tumour characteristics, pre-operative diagnosis and treatment strategies
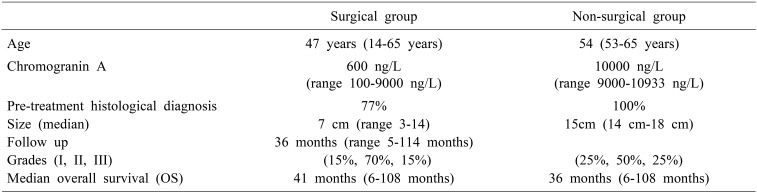
Three patients (23%) had bilobar involvement. Four patients (31%) had multiple lesions whereas the rest had solitary lesions. All patients underwent pre-operative image guided biopsy and pre-treatment histological diagnosis was established in 10 patients (77%). In 3 patients histopathological diagnosis of neuroendocrine tumour could not be established pre-operatively and were reported as adenocarcinoma. However, final histopathological diagnosis of neuroendocrine tumour was established post resection in these patients.
Out of 13 patients who underwent treatment, 8 patients (61%) underwent some form of hepatic resection, in 1 (8%) patient procedure was abandoned in view of underlying cirrhosis and 4 patients (31%) were considered inoperable in view of tumour burden. Out of 8 patients who underwent resection, four patients (50%) underwent TACE (trans-arterial chemoembolization) as a bridge to surgery. In our group TACE was considered in surgical patients with comparatively larger tumours and where surgery waitlist was longer than 4 weeks. Out of 5 (38%) patients including one where surgery was abandoned, 2 (40%) underwent peptide receptor radiotherapy (PRRT) alone, 2 (40%) underwent TACE followed by PRRT and one (20%) underwent trans-arterial radioembolisation (TARE). Liver resections done in 8 patients included 2 right hepatectomies, 2 left extended hepatectomies, 1 right extended hepatectomy, 1 posterior sectionectomy and 2 non-anatomical resections.
Tumour characteristics and histopathological features
All the patients who underwent resection had solitary lesion with median size of 7 cm (range 3–14). Histopathological and immunohistochemistry (IHC) analysis of patients who underwent resection revealed grade I tumour in 1 (13%) patient, Grade II in 5 (62%) patients and Grade III poorly differentiated NEC in 2 (25%) patients. Overall well differentiated NETs Grade I and II was seen in 2 (15%) and 9 (70%) patients respectively, while poorly differentiated NEC was seen in 2 (15%) patients. Mitotic index and Ki67 index was in coherence for all patients.
In our study group, 4 patients were considered inoperable. All patients had bilobar disease with multiple lesions and median cumulative size of 15 cm (14 cm–18 cm). Median Chromogranin A levels were 10000 ng/L (range 9000-10933 ng/L). Well differentiated NETs Grade I and II was seen in 1 (25%) and 3 (75%) patients respectively. Out of 4 (30%) patients, 1 (25%) underwent peptide receptor radiotherapy (PRRT) alone, 2 (50%) underwent TACE followed by PRRT and one (25%) underwent trans-arterial radioembolisation (TARE) (Table 2).
Follow up, recurrence rates and survival analysis
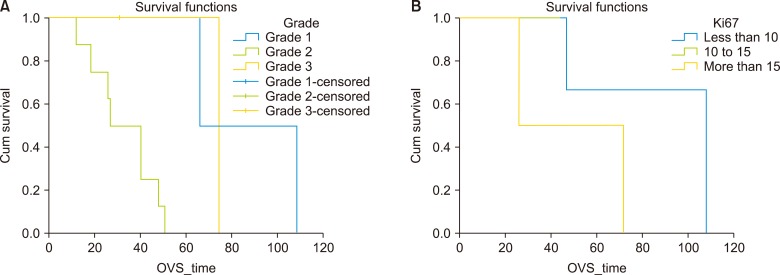
Follow up was conducted in standardised manner as described above and was completed in all patients until October 2018. At a median follow up of 36 months (range 5–114 months), the OS was 92%, with median OS of 47 months (32–61, 95% confidence interval). Median OS in surgical group was 47 months (40–53, 95% confidence interval) that was better but not statistically significant from non-surgical treatment group (36 months). Recurrence was seen in 4 (50%) of resected patients and median recurrence free survival was 20 months (range: 5–72 months). Recurrence was distant in 2 patients (50%) who received systemic chemotherapy in view of poorly differentiated histology, local in 1 (25%) patient who received TACE post-operatively and nodal in 1 (25%) patient who received PRRT post-operatively. On Kaplan Meier survival analysis separation of survival curves was obtained between different grades and Ki 67 indices but difference wasn't statistically significant (Fig. 2).
Go to :
DISCUSSION
PHNETs are extremely rare (less than 0.3% of all neuroendocrine tumours) and hence difficult to diagnose and treat.1 Based on the available literature mean age of presentation is between 4th and 5th decade with no definite gender predominance. In this study mean age was 50 years (range: 14–65 years) with male to female ratio of almost 2:1, however disease distribution was seen in wider age groups, signifying the importance of considering it as a differential in younger age groups also.
Presentation is predominantly non-specific with pain being the most common symptom in 65% of patients and features of carcinoid syndrome being extremely rare. In our series, 38% patients presented with pain and similar percentage of patients presented incidentally, however 24% of patients had diarrhoea and occasional breathlessness which responded to octreotide and represent features of carcinoid syndrome. This is in contrast to findings of others who suggest that carcinoid tumour is an extremely rare presentation of PHNETs.234 The reason for the same is not clear, but functional deficit and insufficient production of serotonin in PHNETs could be among the reasons of low incidence of carcinoid syndrome in PHNETs.
Traditional tumour markers like CA 19-9, AFP and CEA are not raised in majority of patients, however we found that 24% patients had marginally raised CA 19-9 levels but levels were less than 3 times the normal levels and in all patients it returned to normal after treatment. Being functionally deficient, we believe 5-HT and HIAA levels would have very low sensitivity in PHNETs. However, sensitivity and specificity of chromogranin A is high (90 and 92%) and can be used in monitoring disease progress5. Chromogranin A levels were raised in all of our patients and were used to monitor the treatment on follow up with high accuracy in predicting recurrence and response. In our series accuracy of pre-operative biopsy was 77% which is much higher than 57% as reported by Hwang et al.5 Although post resection histopathology serves as the main method of final diagnosis, we are of the opinion that pre intervention diagnosis should be established in all the patients given the high survival potential even in un-resectable group when treated by appropriate non-surgical interventions.6789
The imaging characteristics of PHNETs lack specificity making it difficult to differentiate from other common solid or cystic lesions of the liver.10 Radiographic features are variable and include solid arterially enhancing mass lesion with venous washout, sustained enhancement on delayed scans, hypo-enhancing solid lesions, and hypodense cystic lesions with rim enhancement. In the series by Huang et al.11, authors suggested that cystic changes in imaging could represent distinguishing diagnostic feature of these tumours whenever present. Multidetector Computed Tomography (MDCT) was the predominant cross sectional imaging used in our patients. Findings were variable as described above, with predominant finding being cystic lobulated mass with rim enhancement. We are of the same opinion as Haung et al.11 that multilobulated cystic nature of the lesions in MDCT with areas of haemorrhage may represent a diagnostic finding of PHNETs in appropriate clinical settings.10111213 Both 68Ga-DOTA-TOC (Sensitivity 93%, Specificity 85%) and 68Ga-DOTA-TATE (Sensitivity 96%, Specificity 100%) have high sensitivity and specificity for diagnosing NETs on PET scan. Fusion scan provide information regarding diagnosis, treatment planning, follow-up and aid in ruling out any extra hepatic disease.13 We used 68Ga DOTANOC PET scan in all patients as a modality to rule out extra hepatic disease, primary tumour functional activity and in follow up of treated patients to identify recurrence and latent primary.
Pathological diagnosis is based on both haematoxylin and eosin staining as well as immunohistochemistry was performed using Ki 67, Chromogranin A and Synaptophysin staining. The combination of histo-morphological features in the form of insular, trabecular and glandular as well as IHC features are used to establish the final diagnosis. PHNETs were classified according to WHO (World Health Organisation) 2017 classification.14
Surgical resection is the most preferred treatment modality in these patients. However, it is important to rule out extra hepatic disease or latent primary in all patients by using fusion 68Ga-DOTA-NOCPET scans in all these patients. We use pre-operative TACE as a bridge to surgery in patients with large tumours and where waitlist is expected to be prolonged. Aggressive surgical approach and major hepatectomies for PHNETs is safe and appropriate.15
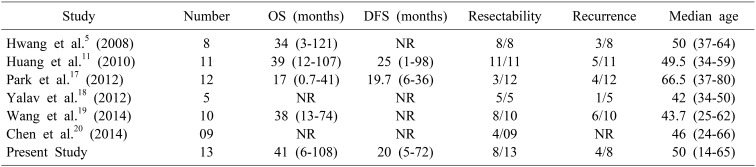
At a median follow up of 36 months (range 5–114 months) we could achieve 92% survival in our study, with median OS of 47 months (32–61, 95% CI). Median OS in the surgical group was 47 months (40–53, 95% CI). In majority of the review series the predominant treatment modality was surgical resection in 84% cases and 5-year survival rates were 78% and 74% respectively in a series by Knox et al.15 and Iwo et al.16 respectively. The recurrence rate of 50% in this series is higher than that 18–20% reported in other series. On survival curve analysis, separation of curves was obtained between different grades and Ki67 indices but was statistically non-significant (Fig. 2). Survival was better in patients with Ki 67 index less than 15%. Hwang et al. in his study found that Ki-67 index is the predictor of prognosis and tumour recurrence.5 In our study there was no extra hepatic involvement, while other studies showed a collective extra hepatic involvement in 19%.13 Overall, surgical resection is effective, safe and prognosis is excellent despite high recurrence rate 8. Table 3 compares variables of major studies on PHNETs with that of present study.
This study has limitations inherent to a retrospective design and an extremely rare disease. Some analytical data may not be representative in view of small sample size. However, this study series does add some vital aspects regarding demography, clinical features, imaging features, treatment options and outcome of this rare disease.
PHNET is an extremely rare NET that predominates in middle-aged population and presents as non-specific abdominal pain or rarely as carcinoid syndrome. Pre-operative diagnosis requires combination of high quality cross-sectional imaging, image guided biopsy, bidirectional endoscopies, tumour markers and fusion scans. Aggressive surgical approach and major hepatectomies for PHNETs is safe and appropriate, while, liver directed therapies may be used as a bridge to resection. In un-resectable and recurrent tumours treatment options include targeted radiotherapy, liver directed therapy, systemic chemotherapy or a combination of these.
Go to :
 XML Download
XML Download