

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The bone is an active tissue that continuously undergoes remodeling through a balance between the resorption and bone formation activities of osteoclasts and osteoblasts [1]. An increase in bone-absorbing osteoclasts over the number of bone-producing osteoblasts escalates an individual's vulnerability to fractures because of the resulting reduced bone quality, osteopenia, or to a greater extent, osteoporosis [23]. Polzer et al. have reported that chronic inflammation is a major risk factor for systemic bone loss leading to osteoporotic fracture. Oxidative stress is frequently associated with such chronic inflammation [4]. In a recent study, administration of a lipopolysaccharide, known as endotoxin, decreased femur mineral content and density in a rat model [5]. Another study affirmed that oxidative stress decreases the quality and quantity of osteoblasts and increases the apoptosis of osteoblasts and osteocytes, further confirming the effect of oxidative stress on increasing bone loss [67].
Bone injury occurs in diverse clinical situations such fracture, orthopedic surgery, plastic gnathoplasty, back surgery, and amputation. During surgery, bone damage and ischemia/reperfusion (I/R) injury often occur and oxygen free radicals are generated. It has been reported that osteoblast-like cells produce considerable amounts of superoxide and hydrogen peroxide (H2O2) during the I/R period [8]. Reactive oxygen species (ROS), including superoxide and H2O2, lead to molecular alteration of cellular components through oxidation of DNA strands, protein cross-links, and side-chains. Consequently, cell morphology, viability, and function may be modified. ROS production is counteracted by an antioxidant defense system including the enzyme scavengers, catalase and glutathione peroxidase [9]. Excessive production of ROS, or a malfunction of the normal cellular redox system, induces oxidative stress, causing cell dysfunction and death [10].
Autophagy has been reported to play an important role in cellular homeostasis by facilitating the survival or death of cells during physiological and pathological conditions [11]. During a state of oxidative stress, autophagy is known to decrease the concentration of ROS and the prevalence of oxidative injuries in order for the cells to maintain energy levels and to continue producing molecules required for cellular survival [12]. However, increased production of ROS shifts their role from inducers of autophagy to a signal for the initiation of mitophagy, a process whereby the mitochondria is removed. This inhibits the anti-oxidative effect of autophagy, and as a result, cell death is accelerated [131415]. On the basis of these reports, we hypothesized that autophagy may be a significant protective factor against oxidative stress.
Remifentanil is an ultra-short acting synthetic µ-opioid receptor agonist, with very rapid onset and offset times owing to its rapid degeneration by nonspecific esterases in the plasma and organs. Owing to these pharmacological properties, remifentanil has been widely applied as an adjuvant analgesic during general anesthesia [16]. Remifentanil pretreatment attenuates hepatic I/R injury in rats via inducible nitric oxide synthase (iNOS). This effect is mediated by a decrease in the production of ROS and an attenuation of the inflammatory response through generation of endogenous NO [17]. Similar suppression of inflammatory responses and inhibition of iNOS expression were observed in mouse models of sepsis [18]. Oxidative stress is closely related to I/R injury. Our previous study indicated that remifentanil preconditioning protected osteoblasts from hypoxia-reoxygenation injury [19]. However, the effects of remifentanil on bone tissue during oxidative stress have not been thoroughly studied. The purpose of our study is to determine whether remifentanil has a protective effect against H2O2-induced oxidative stress in osteoblasts and to examine its influence on factors associated with the proliferation and differentiation of osteoblasts.
MATERIALS AND METHODS
1. Reagents
Remifentanil and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) were both purchased from Sigma (St. Louis, MO, USA). 3-Methyladenine (3-MA, a class III PI3K inhibitor) was obtained from Calbiochem (La Jolla, CA, USA). Antibodies against type I collagen (Col I), BMP-2, osterix, and TGF-β were purchased from Abcam (Cambridge, UK). GAPDH, mouse anti-rabbit IgG antibody, and rabbit anti-mouse IgG antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). All other chemicals and reagents were purchased from Sigma, unless otherwise specified.
2. Cell culture
The human fetal osteoblast cell line (hFOB 1.19) was purchased from ATCC (Rockville, MD, USA). Cells were cultured in Dulbecco's Modified Eagle Medium: Nutrient Mixture F-12 (DMEM/F-12) with 4 mM L-glutamine, 1.5 g/L sodium bicarbonate, 4.5 g/L glucose, and 1.0 mM sodium pyruvate, supplemented with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin (GIBCO-BRL, Rockville, MD, USA). Cells were incubated at 37℃ in a humidified 5% CO2–95% air incubator.
3. Remifentanil treatment
The stock of remifentanil (RPC) (1 ng/ml) was kept frozen at −20℃ until use and was then diluted to the appropriate concentration using DMEM/F-12 when needed. Prior to starting the experiment, cells were grown to approximately 75% confluence and divided into four groups. The control group was incubated at 37℃ in a humidified atmosphere with 5% CO2 for 5 h with no RPC treatment. In the H2O2 group, cells were exposed to 200 µM H2O2 for 2 h and then incubated normally, but were not exposed to remifentanil. In the RPC + H2O2 group, cells were pretreated with 1 ng/ml of remifentanil for 2 h and then exposed to 200 µM H2O2 for 2 h. Lastly, in the 3-MA + RPC + H2O2 group, they were first treated with 1 mM of 3-MA and 1 ng/ml of remifentanil for 1 h and 2 h, respectively. The cells were thereafter exposed to 200 µM of H2O2 for 2 h (Fig. 1).
4. hFOB cell viability assay
The cell viability of hFOB cells was determined using an MTT assay. Cells were cultured in 96-well plates (4 × 103 cells/well). The cells were then treated with the drug concentrations mentioned above. When the treatment was finished, 100 µl of MTT (500 mg/ml) was added to each well. The cells were incubated for 4 h at 37℃. The formazan crystals formed were then solubilized in dimethyl sulfoxide (DMSO) (200 µl/well) by constantly shaking over a period of 15 min. The cell viability was measured using an ELISA reader (Tecan, Männedorf, Switzerland) at an excitatory emission wavelength of 620 nm.
5. Fluorescence microscopy
Cells were harvested and cytocentrifuged onto a clean glass slide. Cells were stained with 1 µg/ml Hoechst 33342 for 15 min at 37℃ in a dark environment and then washed in phosphate-buffered saline (PBS). The slides were mounted using glycerol. The samples were observed and photographed under an epifluorescence microscope (Carl Zeiss, Goettingen, Germany).
6. Flow cytometry analysis
The cells were seeded in 60-mm dishes at 70% confluence and incubated overnight. The remifentanil-treated cells were incubated for varying periods of time according to their conditions . The harvested cells were washed with PBS containing 1% bis (trimethylsilyl) acetamide (BSA) and centrifuged at 2,500 rpm for 10 min. The cells were then re-suspended in ice-cold 95% ethanol with 0.5% Tween 20 to a final concentration of 75% ethanol. After 24 h, the fixed cells were washed in 1% BSA-PBS solution, re-suspended in 1 ml PBS containing 40 µg/ml Ribonuclease A (RNase A) (Sigma), incubated at 4℃ for 30 m, and re-suspended in 10 µg/ml PI solution (Sigma). The DNA contents were examined using a CYTOMICS FC500 flow cytometer (Beckman Coulter ) and the data were analyzed using MultiCycle software, which allowed the simultaneous estimation of cell-cycle parameters and apoptosis.
7. Alizarin red S staining
Mineralization of hFOB cells was determined in 24-well plates using Alizarin red S (Sigma, St. Louis, MO, USA) staining. The cells were washed once with distilled water (DW) after fixation with 4% paraformaldehyde (PFA) and stained with 2% alizarin red solution according to the manufacturer's protocol. For quantification, absorbance of the released alizarin red was measured at 550 nm using a microplate reader.
8. Western blot analysis
The cells (1.5 × 106) were washed twice in ice cold PBS, re-suspended in 200 µl ice cold solubilizing buffer (300 mM NaCl, 50 mM Tris-Cl (pH 7.6), 0.5% Triton X-100, 2 mM phenylmethylsulfonyl fluoride (PMSF), 2 µl/ml aprotinin, and 2 µl/ml leupeptin) and incubated at 4℃ for 1 h. The lysates were centrifuged at 13,200 rpm for 30 min at 4℃. Protein concentrations of cell lysates were determined using a Bradford protein assay (Bio-Rad, Richmond, CA, USA), and 20 µg of protein was resolved using 10% sodium dodecyl sulfate (SDS)/polyacrylamide gel electrophoresis (PAGE) gel. The gel was transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA, USA). After blocking, the membranes were reacted with the appropriate primary antibodies. Immunostaining with secondary antibodies was completed and SuperSignal West Femto substrates (Pierce, Rockford, IL, USA) were used for detection.
RESULTS
Viability of hFOB cells was examined following treatment with remifentanil and H2O2 by measuring MTT reduction. The effects of remifentanil at various concentrations (0, 0.1, 0.5, 1, 2 ng/ml) on cell viability did not significantly differ (Fig. 2A). Pretreatment with 3-MA prior to remifentanil administration had no influence on cell viability (Fig. 2B). The application of H2O2 to hFOB cells at various concentrations (0, 25, 50, 100, 200, 400 µM) for 2 h resulted in decreased cell viability at concentrations of 100 µM or higher (Fig. 2C). Compared to the control group, the H2O2 group showed reduced cell viability (P < 0.05), which was improved by remifentanil pretreatment (1 ng/ml). Treatment with 3-MA (1 mM) decreased the viability of hFOB cells significantly (P < 0.05; Fig. 3).
To assess apoptotic cell death, Hoechst 33342 staining was performed and the cells were viewed under a fluorescence microscope (× 400) to evaluate morphological changes of the nuclei (Fig. 4A). As shown in Fig. 4A, the majority of hFOB cells in the control group show normal morphology with round regular nuclei. In contrast, apoptotic nuclei were observed in the H2O2 group and the 3-MA + RPC + H2O2 group. Remifentanil pretreatment protected hFOB cells from apoptotic cell death during H2O2-induced oxidative stress. Further examination of apoptotic cell death using a fluorescence-activated cell sorter (FACS) showed H2O2-induced apoptosis in 10% of cells in the H2O2 group, compared to the reduced rate of 6.2% in the RPC + H2O2 group (Fig. 4B). This suggests that remifentanil pretreatment attenuates the H2O2-induced apoptosis of hFOB cells. However, pretreatment with 3-MA inhibited the protective effect of remifentanil on cell apoptosis.
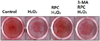
Alizarin red S staining and quantification confirmed the formation of a mineralized matrix after 14 days of differentiation. As shown in Fig. 5, formation of mineralized matrix decreased in the H2O2 group compared to that in the control group. The RPC + H2O2 group showed abundant mineralized matrix compared to the H2O2 group. However, pretreatment with 3-MA counteracted the positive effects of remifentanil and inhibited mineralized matrix formation (Fig. 5).
Expression of bone-related genes such as Col I, BMP-2, osterix, and TGF-β was examined using western blot analysis. H2O2-induced oxidative stress reduced the expression of Col I, BMP-2, and osterix. The negative effects of H2O2 were reversed by remifentanil pretreatment and the expression of Col I, BMP-2, and osterix increased (Fig. 6). TGF-β expression was not inhibited by H2O2 treatment, but significantly increased after remifentanil pretreatment. The expression of all bone-related genes was suppressed by pretreatment with 3-MA. These results suggest that remifentanil pretreatment induces the expression of Col I, BMP-2, osterix, and TGF-β in hFOB cells under H2O2-induced oxidative stress.
DISCUSSION
In this study, we investigated whether remifentanil pretreatment protects hFOB cells against H2O2-induced oxidative stress. We demonstrated that remifentanil increased the differentiation of osteoblasts and supported matrix formation following exposure to oxidative stress. Pretreatment with 3-MA, a selective autophagy inhibitor, suppressed the anti-oxidant effects of remifentanil. Therefore, our study serves as the evidence for the involvement of autophagy in remifentanil's protective effect against oxidative stress. This stress leads to cytotoxicity through ROS overproduction, which harms cardinal cellular components, such as proteins, lipids, and DNA, owing to their highly reactive properties [20]. For example, hydroxyl radicals—a type of ROS—generate oxidative injuries, such as oxidized bases, abasic sites, si ngle-strand breaks, double-strand breaks, and DNA-protein crosslinks, causing direct damage to the DNA backbone [2122]. In addition, oxidative stress is responsible for the pathophysiology of the aging process and may play a crucial role in the pathogenesis of atherosclerosis, neurodegenerative disease, cancer, and diabetes [23]. Therefore, the development of therapeutic options to prevent oxidative stress and its consequential damage is crucial.
Osteoporosis is influenced by various factors including nutrition, hormones, cytokines, and aging. Indeed, according to a recent study, ROS may be involved in the pathogenesis of bone loss and are responsible for the development of osteoporosis [7]. It has already been reported that oxidative stress modulates the differentiation and survival of osteoblasts and decreases the overall level of bone formation. In addition, oxidative stress promotes osteoclastogenesis and bone resorption, especially in aged people [2425]. With life expectancy generally increasing, the probability of undergoing surgery at some point in one's lifetime is increasing as well [26]. In particular, fractures following traumatic events such as a fall or slip are common in elderly, osteoporotic patients who then require surgery [27]. During surgery, oxidative stress increases due to I/R injury. Elderly patients are vulnerable to oxidative stress due to a deterioration of antioxidant defenses, leading to a delay in bone healing [28]. Our result is both novel and useful because it is the first study that shows the protective effects of remifentanil against oxidative stress in osteoblasts.
In our study, we observed that remifentanil pretreatment increased the synthesis and expression of bone-related proteins, such as Col I, BMP-2, osterix, and TGF-β. Col I is the major structural protein in the extracellular matrix of bone, accounting for 90% of bone matrix proteins, and playing a crucial role in osteoblast cell adhesion, proliferation, and differentiation [29]. Osterix is a transcription factor that regulates important osteoblast genes such as Col I, osteocalcin, and osteopontin [30]. BMPs are an important subclass of growth factors within the TGF-β superfamily. In particular, BMP-2 plays a critical role in bone healing. BMP-2 and TGF-β have important functions in inducing osteoblast differentiation and bone formation during the preosteoblast stage [3132]. This is the first study reporting the effects of remifentanil on factors associated with the proliferation and differentiation of hFOB cells in H2O2-induced oxidative stress conditions.
To investigate the degree to which autophagy may be involved in remifentanil's reduction of H2O2-induced oxidative stress in hFOB cells, we added 3-MA prior to remifentanil pretreatment. 3-MA inhibits autophagy by blocking autophagosome formation via the inhibition of class III phosphatidylinositol 3-kinases. Autophagy is generally considered a cell survival mechanism. However, depending on the level of cellular stress, it may serve the opposite function. Autophagy has been reported to be a cell death mechanism causing autophagic cell death or type II programmed cell death [3334]. In our study, pretreatment with 3-MA inhibited the protective influence of remifentanil against H2O2-induced oxidative stress and its effects on cell viability, apoptosis, mineralized matrix formation, and expressions of bone-related genes. These results suggest that the protective effect of remifentanil against oxidative stress is mediated by autophagy. However, there is still was insufficient evidence to conclusively state that the all antioxidant effects of remifentanil pretreatment are mediated solely by autophagy. Additional studies are required to verify this uncertainty.
The results of the present study revealed that remifentanil pretreatment has a protective effect on H2O2-induced hFOB cells, increasing differentiation of osteoblasts and expression of bone-related genes. We demonstrated the possibility that autophagy is involved in the protective mechanism of remifentanil in response to H2O2-induced oxidative stress. Although further research is required to identify the full effect of remifentanil on oxidative injury, our study provides foundational evidence that remifentanil reduces oxidative damage in hFOB cells. For correlation and translation to clinical situations, such as osteoporosis and fracture, further clinical studies are required





 XML Download
XML Download