

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Oxidative stress is an imbalance between oxidant molecules and antioxidant species that protect cells from harmful effects of oxidants. The major molecules that are produced as a result of oxidative stress are reactive free radicals [1]. Reactive oxygen species (ROS), one of the major classes of reactive free radicals, are produced during aerobic metabolism in all oxygenic organisms. Cells also generate ROS as signaling molecules through the activation of various oxidases and peroxidases [23]. However, when ROS are present in excess, they have a direct oxidizing effect on crucial cell components needed for survival, such as lipids, proteins, and DNA, therefore ROS are involved in cell injury, necrosis, and apoptosis, which are often associated with human diseases [4567]. ROS can be produced in the lung or other organs as a consequence of high oxygen therapy or hypoxia-reperfusion, and they can stimulate cell death pathways associated with tissue damage [8].
Autophagy is a degradation system within cells resulting in the reuse of intracellular proteins or other damaged organelles by nonselective, bulky engulfment, and digestion of cellular components into reusable molecules [910]. In general, autophagy is thought to be induced under stressful circumstances such as oxidative stress, starvation, and hypoxia-reoxygenation [11]. ROS accumulation induces autophagy, which then serves to reduce ROS levels [12].
Remifentanil is an ultra-short-acting, selective muopioid receptor agonist that has attracted attention because of its esterase-based metabolism, minimal accumulation, and very rapid onset and offset of clinical action [13141516]. Like several other anesthetics, remifentanil has been reported to protect various organs against ischemia-reperfusion injury (IRI) such as myocardium, brain, kidney and liver, possibly by inhibiting oxidative stress responses [17181920]. Autophagy is induced earlier than apoptosis so it can protect cells against damage in situations such as IRI [10]. However, the effects of remifentanil on Cos-7 cell survival and autophagy during oxidative stress has not been examined. Therefore, this study investigated whether remifentanil pretreatment has a protective effect on Cos-7 cells exposed to oxidative stress, and we determined the influence of this compound on intracellular autophagy and apoptotic cell death.
MATERIALS AND METHODS
1. Reagents
Remifentanil (Ultiva™ inj., 1 mg vial, GlaxoSmithKline, Belgium) was diluted in dimethyl sulfoxide (DMSO). Hoechst 33342 was purchased from Sigma. 3-[4,5-dimethylthiazol-2-yl]2,5-diphenyl tetrazolium bromide (MTT), acridine orange (AO), monodansylcadaverine (MDC), and 3-methyladenine (3-MA, class III PI3K inhibitor) were obtained from Calbiochem (La Jolla, CA, USA). Antibodies used in the study were as follows: LC3-II (microtubule-associated protein 1 light chain 3 form II) (1:3,000) and Beclin-1 (1:1,000) were obtained from Abcam, p62 (1:1,000) and Atg5 (1:500) from Santa Cruz. Secondary antibodies against rabbit (1:3,000), and mouse (1:3,000), immunoglobulins were purchased from Bio-Rad.
2. Cell culture
Cos-7 cells were obtained from the American Type Culture Collection (ATCC, Manassas, USA). Cells were cultured in Dulbecco's modified Eagle medium (DMEM, GIBCO) supplemented with 10% inactivated fetal bovine serum (FBS, GIBCO) containing 500 µg/ml penicillin and 500 µg/ml streptomycin (GIBCO), and cells were incubated at 37℃ in a humidified atmosphere with 5% CO2. Media were changed every 3 days.
3. Remifentanil treatment
Remifentanil solutions in DMSO were kept frozen at −4℃ until use. The stock was diluted to the appropriate concentration in DMEM when needed. Prior to remifentanil treatment, cells were grown to about 80% confluence and then exposed to remifentanil at different concentrations (0, 0.1, 0.5, 1, 2 ng/ml) for 24 h. Cells grown in medium containing an equivalent amount of DMSO without remifentanil served as a control. Cells were randomly divided into the following groups: (1) Control: cells were incubated in normoxia (5% CO2, 21% O2, and 74% N2) without remifentanil treatment. (2) H2O2: cells were exposed to H2O2 (0, 50, 100, 200, or 400 concentrations of H2O2) without remifentanil treatment for 24 h. (3) RPC + H2O2: cells pretreated with remifentanil were exposed to H2O2 for 24 h. (4) 3-MA + RPC + H2O2: cells pretreated with 3-MA and remifentanil were exposed to H2O2 for 24 h.
4. MTT assay
Cell viability was measured using a quantitative colorimetric assay with thiazolyl blue tetrazolium bromide (MTT, AMRESCO), which shows the mitochondrial activity of living cells. Cos-7 cells were seeded in 96-well plates. After drug treatment as indicated, cells were incubated with 300 µl MTT reagent (final concentration 0.5 mg/ml) for 1.5 h at 37℃. The reaction was terminated by adding 200 µl DMSO. Cell viability was measured by an ELISA reader (Tecan, Mannedorf, Switzerland) at 570 nm excitatory emission wavelength.
5. Hoechst staining (with 1 ng/ml remifentanil treatment)
After remifentanil treatment, the cells were transferred onto a clean, fat-free glass slide using a cytocentrifuge. They were stained with 4 µg/ml Hoechst 33342 for 10 min at 37℃ in darkness and washed twice in PBS. The slides were mounted with glycerol. The samples were observed and photographed using epifluorescence microscopy (Carl Zeiss, Göettingen, Germany). The number of cells with condensed or fragmented nuclei was determined by a blinded observer from a random sampling of 3 × 102 cells per experiment. Three independent experiments were conducted.
6. Fluorescence microscopy
Cells were grown on coverslips and treated with remifentanil. After 24 h, cells were stained with 0.05 mM MDC, a selective fluorescent marker for autophagic vacuoles, at 37℃ for 1 h. Changes in cellular fluorescence were observed using a fluorescence microscope (Axioskop, Carl Zeiss, Germany). For further detection of the acidic cellular compartment, we used acridine orange (AO), which emits bright red fluorescence in acidic vesicles but green fluorescence in the cytoplasm and nucleus. Cells were stained with 1 µg/ml AO for 15 min and washed with PBS. Autophagic vacuole organelle (AVO) formation was determined using a confocal microscope LSM 700 (Carl Zeiss, Germany).
7. Western blot analysis
Cells (2 × 106) were washed twice in ice-cold PBS, resuspended in 200 µl ice-cold solubilizing buffer (300 mM NaCl, 50 mM Tris-Cl [pH 7.6], 0.5% Triton X-100, 2 mM PMSF, 2 µl/ml aprotinin and 2 µl/ml leupeptin) and incubated at 4℃ for 30 min. The lysates were centrifuged at 14,000 rpm for 15 min at 4℃. Cell lysate protein concentrations were determined with a Bradford protein assay (Bio-Rad, Richmond, CA, USA) and 20 µg of total proteins were resolved by 10% SDS-PAGE. Proteins were transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA, USA) and reacted with the appropriate primary antibody. Immunostaining with secondary antibodies was detected using SuperSignal West Femto (Pierce, Rockford, IL, USA) enhanced chemiluminescence substrate and observed with an Alpha Imager HP (Alpha Innotech, Santa Clara, USA).
8. Statistical analysis
Three independent experiments were performed for each experimental group and each experiment was performed in triplicate. The significance of differences between experimental and control groups were determined (p < 0.01 and 0.05) using a paired Student's t-test using SPSS for Win 12.0. Fluorescence activated cell sorting (FACS) analysis of Cos-7 cells was also performed for control, H2O2, RPC + H2O2, and 3-MA + RPC + H2O2 groups to determine apoptotic cell populations of each group.
RESULTS
1. Remifentanil pretreatment increased the viability of Cos-7 cells after oxidative stress
The effect of remifentanil on Cos-7 cells was investigated over a wide concentration range (0–2 ng/ml). Remifentanil and 3-MA treatment did not result in any significant effect on the viability of Cos-7 cells (Fig. 1A), while the cell viability in H2O2-treated groups decreased in a concentration-dependent manner (Fig. 1B). The viability of cells in the RPC + H2O2 group was 92.1% relative to the control cells (100%), while in the H2O2 group, viability was 67.2% (Fig. 2). In addition, the 3-MA + RPC + H2O2 group showed 75.8% cell viability, which was lower than cells pretreated with remifentanil before oxidative stress, but higher than cells treated with H2O2 alone (Fig. 2).
2. Oxidative stress-dependent apoptosis was suppressed by remifentanil
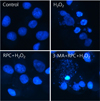
The effect of remifentanil on apoptosis was examined by Hoechst 33342 staining of Cos-7 cells exposed to normoxia (control), H2O2, RPC + H2O2, and 3-MA + RPC + H2O2 (Fig. 3). Cells were examined under a fluorescence microscope (400 ×). The majority of Cos-7 cells in the control and RPC + H2O2 groups showed normal morphology with round, regular nuclei. On the other hand, apoptotic bodies were seen in the H2O2 group and the 3-MA + RPC + H2O2 group. Pretreating with remifentanil effectively reduced Cos-7 cell apoptosis as observed by their normal morphology. In addition, FACS analysis of Cos-7 cells showed that the percentage of apoptotic cells in the remifentanil-preconditioned group was 6.4%, while in oxidative stressed group the percentage of apoptotic cells was 10.3% (Fig. 4).
3. Remifentanil pretreatment leads to autophagy induction in Cos-7 cells
A remarkable accumulation of autophagy-specific staining by MDC was observed around the nuclei in the RPC + H2O2 group of Cos-7 cells (Fig. 5A). Similarly, AO staining indicated by red fluorescent spots appeared in the RPC + H2O2 group cells, while the control, H2O2, and 3-MA + RPC + H2O2 groups showed mainly green cytoplasmic fluorescence (Fig. 5B). We clarified the role of remifentanil-induced autophagy in Cos-7 cells by investigating the consequences of treating with 3-MA, a selective autophagy inhibitor. The inhibitory effects on AVO formation by 3-MA was confirmed by quantitatively measuring the red fluorescence ratio after AO staining. We examined the activation of autophagy related proteins in each group of cells by western blotting. The recruitment of LC3-II to the membrane occurs via an Atg5-dependent mechanism, and thus Atg5 is essential for autophagosome formation in vivo. The levels of Atg5 and Beclin-1, LC3-II, and p62 were decreased or similar in the oxidative stressed group compared to the control group, but were increased in the RPC + H2O2 group (Fig. 6). In addition, the levels of Atg5, Beclin-1, LC3-II, and p62 were lowest in the 3-MA + RPC + H2O2 group. Levels of these autophagy-related proteins were increased when autophagy was induced by remifentanil, and they were decreased when autophagy was suppressed by 3-MA.
DISCUSSION
The purpose of this study was to determine whether remifentanil pretreatment exhibits protective effects on Cos-7 cells undergoing oxidative stress, and to determine the influence of this compound on intracellular autophagy and apoptotic cell death. The present study showed several principal findings. First, we found that remifentanil pretreatment increased the viability of cells exposed to oxidative stress (Fig. 2). Importantly, remifentanil pretreatment did not affect cell viability under normal conditions, nor did treatment with 3-MA (Fig. 1A). Second, oxidative stress-dependent apoptosis was suppressed by remifentanil pretreatment. We found that the number of apoptotic bodies was reduced in remifentanil-preconditioned cells (Fig. 3), and the apoptotic cell percentage in remifentanil-preconditioned Cos-7 cells under oxidative stress was lowered by 6.4% compared to cells exposed to oxidative stress without remifentanil pretreatment (Fig. 4). Third, remifentanil pretreatment protected Cos-7 cells against oxidative stress by activating intracellular autophagy, which was likely the anti-apoptotic mechanism. Western blot analysis showed that remifentanil pretreatment upregulated protein levels of ATG5, Beclin-1, LC3-II, and p62, which are autophagy-related proteins (Fig. 6) [21]. Lastly, as seen by autophagy-specific staining using MDC and AO, the RPC + H2O2 group showed the presence of autophagosomes by the observation of blue dots (MDC) or red spots (AO), and the autophagy pathway-specific inhibitor 3-MA blocked autophagosome formation (Fig. 5). These results regarding autophagy might be related with the cell viability finding (Fig. 2) that cells exposed to oxidative stress alone showed 67.2% cell viability, while cells under oxidative stress with remifentanil preconditioning showed 92.1% cell viability. These results show that remifentanil preconditioning has a protective effect on cells exposed to oxidative stress. The activation of autophagy is the mechanism by which remifentanil protects cells undergoing oxidative stress. Remifentanil preconditioning induced the autophagy response and it resulted in decreased cell death upon exposure to oxidative stress.
Autophagy is a cellular process used to deliver cytoplasmic organelles to the lysosome for degradation. It is most often implicated in cell survival by cleaning up damaged organelles and harmful substances inside the cell, but also by providing building blocks and nutrients to the cell [2223]. However, autophagy has also been related to cell death [22232425]. Recent studies suggest that autophagy has a great effect on determining cell fate toward survival or death, depending on the stimulus, cell type, and context [92426]. In this study, we assumed that cell survival would be promoted if autophagy is activated in the early period of cellular oxidative stress by preconditioning with remifentanil.
Oxidative stress can lead to cell death through the production of ROS, one of the major reactive free radical species induced by oxidative stress [127]. ROS can cause cell death by necrotic or apoptotic pathways due to its direct oxidizing effects on macromolecules such as lipids, proteins, and DNA [14828], and ROS are involved in many diseases including cardiovascular diseases, nephropathy, neurodegenerative diseases, and aging [1672829]. Previous studies demonstrate an essential role for ROS in the activation of autophagy, which, in turn, leads to reduced ROS levels [3031]. The results of this study indicate that remifentanil preconditioning suppresses cell damage and apoptosis due to oxidative stress, and increases cell viability by upregulating autophagy.
There have been various studies regarding the protective effect of remifentanil and its mechanisms against oxidative stress, similar to the effects of other types of anesthetics. Ge Zhao et al. (2013) showed that remifentanil may protect hepatocytes against IRI by reducing hepatic cell apoptosis [32]. T.Z. Zhang et al. (2014) showed that high-dosage remifentanil preconditioning played a protective role on brain cells, possibly by inhibiting the oxidative stress response [20]. In addition, Zhang Y et al. (2005) concluded that cardiac delta and kappa opioid receptors, but not mu opioid receptors, mediate cardioprotection by remifentanil preconditioning on ischemia-reperfused rat hearts [183334], and H.S. Kim et al. (2009) suggested that the cardioprotective effect of remifentanil resulted from preserving anti-apoptotic pathways in ischemia-reperfused rat hearts, regardless of the administration period [19]. Cho SS et al. (2013) showed that preconditioning with remifentanil altered intestinal IRI and systemic inflammatory responses in mice, therefore they concluded that prophylaxis with remifentanil may be valuable in various clinical conditions of human IRI [17]. Recent mechanistic studies revealed a protective effect of opioids against oxidative stress, with opioid receptors as the major contributors to this effect [333435]. However, it was not known if the protective effect of remifentanil was related to autophagy in cells under oxidative stress; hence, we sought to examine this possibility in the current study.
There are several limitations in present study. First, this study was performed in vitro. Further studies are needed to determine if the protective effect of remifentanil occurs through activation of autophagy in human cells and organs under oxidative stress. Secondly, the protective effect of remifentanil seemed to be through other pathways as well as activation of autophagy. When autophagy activation was suppressed by treating with 3-MA in the remifentanil-preconditioned Cos-7 cells under oxidative stress, cell viability was 75.8%, which was lower than the cell viability of 92.1% observed in the RPC + H2O2 group, but still higher than the H2O2 group (Fig. 2). There might be other remifentanil-mediated pathways affecting cell viability under oxidative stress, and further studies regarding this are needed. Third, further studies should be conducted to determine the mechanisms by which remifentanil increases autophagy activity in cells exposed to oxidative stress.
The current study showed that remifentanil preconditioning stimulated autophagy and increased cell viability in the oxidative stressed model of Cos-7 cells. In addition, remifentanil did not affect cell viability in cells not exposed to oxidative stress. Therefore, we suggest that cell autophagy in was activated only under oxidative stress conditions, and remifentanil preconditioning increased cell survival by activating autophagy.





 XML Download
XML Download