

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Idiopathic pulmonary fibrosis (IPF) is a specific form of progressive, irreversible, and fatal fibrosing interstitial pneumonia of unknown etiology.1 Although two antifibrotic agents, nintedanib and pirfenidone, are available to reduce disease progression in patients with mild to moderate IPF, randomized controlled trials (RCTs) have not demonstrated significant improvement in the IPF mortality rate or acute exacerbations,2 indicating a need for novel or additional agents to treat IPF.
Pulmonary hypertension (PH) is common in IPF patients.3 The presence of PH in IPF, as measured by Doppler echocardiography, ranges widely from 32% to 85%, depending on the disease severity, evaluation method, and diagnostic criteria.456789 Furthermore, PH has been observed in approximately 20% of patients with mild to moderate restricted IPF,10 and in 28.8%–46.1% of patients with end-stage IPF awaiting lung transplantation, as measured by right heart catheterization.4711 The presence of PH is correlated with mortality, impaired gas exchange, and reduced exercise capacity in IPF patients,10 and it has been well established that the development of PH plays an important role in IPF prognosis.1213 Thus, active intervention for IPF-associated PH might improve long-term outcomes, and it could be plausible to consider PH-specific agents as a therapeutic option for IPF patients.
Current international guidelines for the diagnosis and management of IPF do not recommend PH-specific agents for the majority of IPF patients.3 However, several previously published trials have reported that they offer clinical benefits for exercise capacity, symptoms, and quality of life.141516 Considering the contradictory results published about PH-specific agents, we conducted a systematic review and meta-analysis to evaluate the clinical efficacy and safety of PH-specific agents for IPF patients.
METHODS
Data sources and search strategy
We performed electronic literature searches of MEDLINE, EMBASE, and the Cochrane Central Register using search filters provided by SIGN (http://www.sign.ac.uk/methodology/filters.html) and following the recommended guidelines of the Preferred Reporting Items for Systematic Reviews and Meta-Analyses.17 Searches were limited to human studies without language restriction through November 2018. As search terms, we used the following keywords: “idiopathic pulmonary fibrosis” or “fibrosing alveolitis” or “usual interstitial pneumonia” AND “endothelin receptor antagonist” or “prostacyclin” or “prostacyclin derivative” or “phosphodiesterase-5 inhibitor” or “soluble guanylate cyclase stimulators” or “bosentan” or “ambrisentan” or “sitaxentan” or “macitentan” or “sildenafil” or “tadalafil” or “udenafil” or “vardenafil” or “riociguat” or “epoprostenol” or “treprostinil” or “beraprost” or “iloprost.” We also performed a manual search of the references cited in relevant review articles.
Study selection and data extraction
The following predetermined inclusion criteria were used: 1) full-length reports published in peer-reviewed journals; 2) RCTs comparing PH-specific agents with controls; 3) active interventions for one of the four PH-specific agent classes: endothelin receptor antagonists (ERAs), prostacyclin or prostacyclin analogues, soluble guanylate cyclase stimulators (sGCSs), or phosphodiesterase type 5 (PDE5) inhibitors; 4) patients diagnosed with IPF according to international consensus guidelines1819; and 5) clinical outcomes and adverse events data available.
Two investigators independently retrieved potentially relevant studies, reviewed each study according to the predefined eligibility criteria, and extracted data. Disagreements were resolved through discussion between them. For each study, a predesigned form was used to extract data on the authors, publication year, population, interventions, controls, clinical outcomes, and adverse events. We excluded case reports, case series, letters to the editor, editorials, and commentaries because the methods and results could not be fully analyzed. The primary outcome of interest was all-cause mortality to end of study, defined as the time from random assignment to death from any cause. Secondary outcomes were forced vital capacity (FVC), diffusion capacity of the lung for carbon monoxide (DLco), 6-minute walk distance test (6MWD), St. George Respiratory Questionnaire (SGRQ) total score, Borg dyspnea score after walk test, and serious adverse events related to treatment.
Risk of bias assessment
The quality of the RCTs was assessed using the Cochrane Handbook for Systematic Reviews of Interventions' risk of bias tool.20 This scale evaluates the following criteria: sequence generation/allocation concealment (selection bias), blinding of participants and personnel (performance bias), blinding of outcome assessment (detection bias), incomplete outcome data (attrition bias), selective outcome reporting (reporting bias), and other sources of bias. Risk of bias was labeled as high, low, or unclear. Reviewer agreement was achieved through consensus. If any randomized or blinded item was judged as high risk, the trial was assigned a high risk of bias. The publication bias was assessed using Begg's and Egger's regression tests.2122
Data synthesis and statistical analysis
Summary measures of risk ratios (RRs) or hazard ratios (HRs) were calculated as appropriate with 95% confidence intervals (CIs) to assess the treatment efficacy of PH-specific agents. Statistical heterogeneity was assessed by calculating I2 values to quantify the degree of heterogeneity between studies23 on a scale of 0%–100%. We considered a value of I2 between 30% and 60% as an indication of moderate heterogeneity and > 60% as significant between-study heterogeneity.23 If substantial heterogeneity existed, a random effect model was used for the analysis; otherwise, a fixed effect model was used. We also conducted sensitivity analyses using the leave-one-out method to evaluate the influence of highly heterogeneous individual studies on the overall effect estimate. Subgroup analyses for the primary outcome of interest were planned based on the type of PH-specific agent, sample size, age, FVC, DLco, and the extent of honeycombing.
A P value < 0.05 was considered statistically significant. We analyzed data using Review Manager Software, version 5.3 (The Nordic Cochrane Centre, The Cochrane Collaboration, Copenhagen, Denmark) and Stata version 14.2 (StataCorp LP, College Station, TX, USA).
RESULTS
Study search, characteristics of included studies, and study quality
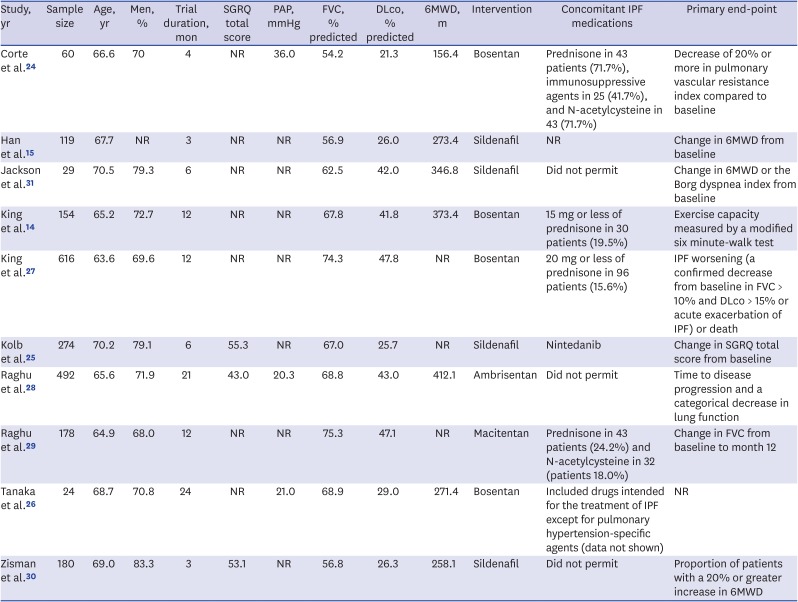
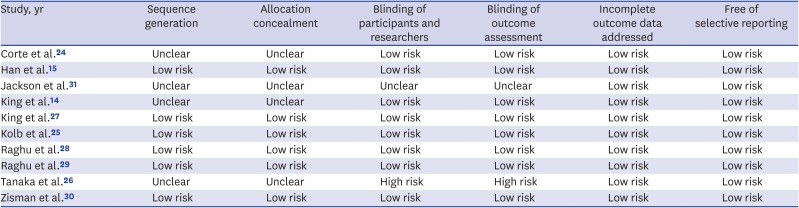
The electronic database search yielded 579 published articles (Fig. 1). After removal of duplicate articles, the titles and abstracts of 488 references were screened. Of these, 43 eligible articles were selected. After the full-text review, 10 studies reported at least one primary or secondary outcome that could be combined in this meta-analysis.14152425262728293031 Individual characteristics of the selected studies are shown in Table 1. All included articles were published between 2008 and 2018. The number of patients in the trials ranged from 24 to 616. The active interventions were ERAs in six trials (bosentan in four, ambrisentan in one, and macitentan in one) and a PDE5 inhibitor in four trials (sildenafil in all). One study included idiopathic fibrotic nonspecific interstitial pneumonia as well as IPF,24 and another study included results from a combined therapy of nintedanib and sildenafil.25 The results from the quality assessment of the included studies are shown in Table 2. One trial was judged to be at high risk of bias because it did not blind participants and researchers, nor did it blind the outcome assessment.26
Table 1
Main characteristics of the randomized controlled trials included in the meta-analysis
| Study, yr | Sample size | Age, yr | Men, % | Trial duration, mon | SGRQ total score | PAP, mmHg | FVC, % predicted | DLco, % predicted | 6MWD, m | Intervention | Concomitant IPF medications | Primary end-point |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Corte et al.24 | 60 | 66.6 | 70 | 4 | NR | 36.0 | 54.2 | 21.3 | 156.4 | Bosentan | Prednisone in 43 patients (71.7%), immunosuppressive agents in 25 (41.7%), and N-acetylcysteine in 43 (71.7%) | Decrease of 20% or more in pulmonary vascular resistance index compared to baseline |
| Han et al.15 | 119 | 67.7 | NR | 3 | NR | NR | 56.9 | 26.0 | 273.4 | Sildenafil | NR | Change in 6MWD from baseline |
| Jackson et al.31 | 29 | 70.5 | 79.3 | 6 | NR | NR | 62.5 | 42.0 | 346.8 | Sildenafil | Did not permit | Change in 6MWD or the Borg dyspnea index from baseline |
| King et al.14 | 154 | 65.2 | 72.7 | 12 | NR | NR | 67.8 | 41.8 | 373.4 | Bosentan | 15 mg or less of prednisone in 30 patients (19.5%) | Exercise capacity measured by a modified six minute-walk test |
| King et al.27 | 616 | 63.6 | 69.6 | 12 | NR | NR | 74.3 | 47.8 | NR | Bosentan | 20 mg or less of prednisone in 96 patients (15.6%) | IPF worsening (a confirmed decrease from baseline in FVC > 10% and DLco > 15% or acute exacerbation of IPF) or death |
| Kolb et al.25 | 274 | 70.2 | 79.1 | 6 | 55.3 | NR | 67.0 | 25.7 | NR | Sildenafil | Nintedanib | Change in SGRQ total score from baseline |
| Raghu et al.28 | 492 | 65.6 | 71.9 | 21 | 43.0 | 20.3 | 68.8 | 43.0 | 412.1 | Ambrisentan | Did not permit | Time to disease progression and a categorical decrease in lung function |
| Raghu et al.29 | 178 | 64.9 | 68.0 | 12 | NR | NR | 75.3 | 47.1 | NR | Macitentan | Prednisone in 43 patients (24.2%) and N-acetylcysteine in 32 (patients 18.0%) | Change in FVC from baseline to month 12 |
| Tanaka et al.26 | 24 | 68.7 | 70.8 | 24 | NR | 21.0 | 68.9 | 29.0 | 271.4 | Bosentan | Included drugs intended for the treatment of IPF except for pulmonary hypertension-specific agents (data not shown) | NR |
| Zisman et al.30 | 180 | 69.0 | 83.3 | 3 | 53.1 | NR | 56.8 | 26.3 | 258.1 | Sildenafil | Did not permit | Proportion of patients with a 20% or greater increase in 6MWD |
Data are presented as number or mean, unless otherwise stated.
6MWD = 6-minute walk distance test, DLco = diffusion capacity of the lungs for carbon monoxide, FVC = forced vital capacity, IPF = idiopathic pulmonary fibrosis, NR = not reported, PAP = pulmonary arterial pressure, SGRQ = St. George's Respiratory Questionnaire.
Table 2
Risk of bias among the studies included in the meta-analysis
| Study, yr | Sequence generation | Allocation concealment | Blinding of participants and researchers | Blinding of outcome assessment | Incomplete outcome data addressed | Free of selective reporting |
|---|---|---|---|---|---|---|
| Corte et al.24 | Unclear | Unclear | Low risk | Low risk | Low risk | Low risk |
| Han et al.15 | Low risk | Low risk | Low risk | Low risk | Low risk | Low risk |
| Jackson et al.31 | Unclear | Unclear | Unclear | Unclear | Low risk | Low risk |
| King et al.14 | Unclear | Unclear | Low risk | Low risk | Low risk | Low risk |
| King et al.27 | Low risk | Low risk | Low risk | Low risk | Low risk | Low risk |
| Kolb et al.25 | Low risk | Low risk | Low risk | Low risk | Low risk | Low risk |
| Raghu et al.28 | Low risk | Low risk | Low risk | Low risk | Low risk | Low risk |
| Raghu et al.29 | Low risk | Low risk | Low risk | Low risk | Low risk | Low risk |
| Tanaka et al.26 | Unclear | Unclear | High risk | High risk | Low risk | Low risk |
| Zisman et al.30 | Low risk | Low risk | Low risk | Low risk | Low risk | Low risk |
Overall survival
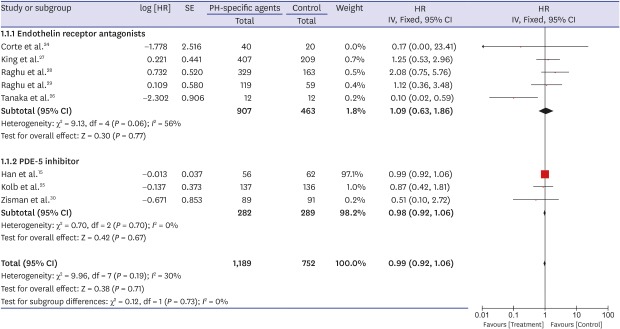
A forest plot of the primary outcome—effects of PH-specific agents on all-cause mortality at the study endpoints—is shown in Fig. 2. Data regarding the effects of PH-specific agents on all-cause mortality to end of study in IPF patients were available for eight trials, and we analyzed them using an inverse variation method.1524252627282930 Of a total of 1,941 enrolled patients, 1,189 were treated with PH-specific agents, and 752 received placebo or no treatment. In pooled estimates, all-cause mortality to end of study did not differ significantly between the PH-specific agent group and the control group (HR, 0.99; 95% CI, 0.92, 1.06; P = 0.71) (Fig. 2). There was a moderate degree of statistical heterogeneity among the eight trials (I2 = 30%; P = 0.19). To investigate the effect of each individual study on the overall estimates, we performed a sensitivity analysis by calculating the pooled HRs while successively excluding one study at a time. One study had a significantly different all-cause mortality estimate than the others.26 Even after excluding that study,26 however, all-cause mortality did not differ significantly between the groups, although the heterogeneity decreased (HR, 0.99; 95% CI, 0.92, 1.06; P = 0.78; I2 = 0%). We found no evidence of publication bias using Begg's (P = 0.266) and Egger's (P = 0.516) tests.
Fig. 2
Pooled effects of PH-specific agents versus controls on overall survival time.
PH = pulmonary hypertension, SE = standard error, HR = hazard ratio, CI = confidence interval.
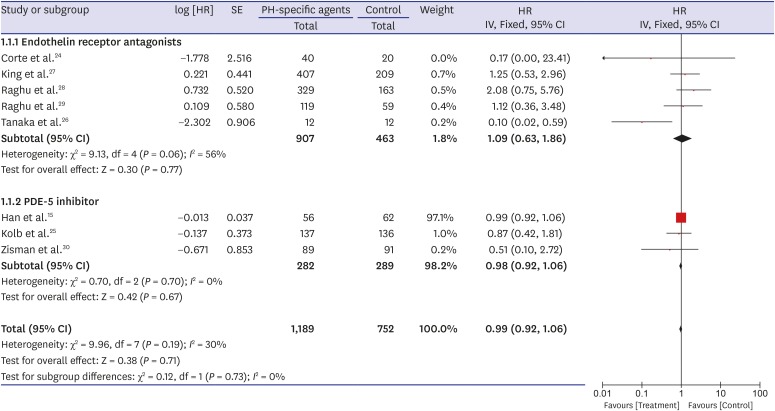
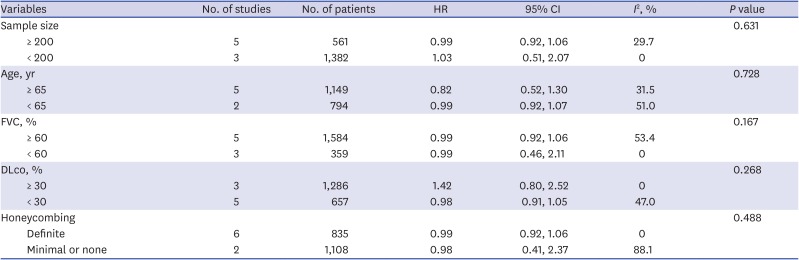
Because the result for the primary outcome was not significant, we performed subgroup analysis. When the analysis was restricted to patients treated with ERAs or PDE-5 inhibitors, all-cause mortality to end of study did not differ significantly between the groups (HR, 1.09; 95% CI, 0.63, 1.86; P = 0.77; I2 = 56% for ERAs and HR, 0.98; 95% CI, 0.92, 1.06; P = 0.67; I2 = 0% for the PDE-5 inhibitor) (Fig. 2). Also, all-cause mortality to end of study did not differ significantly by sample size (≥ 200 vs. < 200), age (≥ 65 vs. 65), mean FVC (≥ 60% vs. 60%), mean DLco (≥ 30% vs. < 30%), or the extent of honeycombing (definite vs. minimal or none) (Table 3).
Table 3
Subgroup analysis of the randomized controlled trials included in the meta-analysis
Lung function
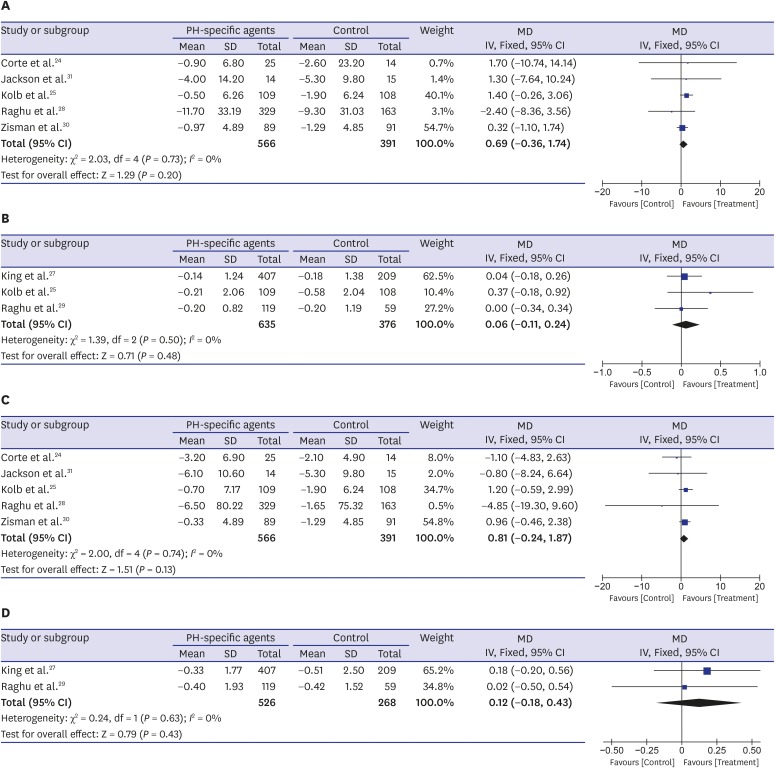
Seven RCTs reported data on changes in FVC from baseline,24252728293031 and it did not differ significantly between the PH-specific agent group and the control group (mean difference [MD], 0.69% predicted; 95% CI, −0.36, 1.74; P = 0.20; I2 = 0% for FVC % predicted and MD, 0.06 L; 95% CI, −0.11, 0.24; P = 0.48; I2 = 0% for FVC L) (Fig. 3A and B). Data regarding DLco changes from baseline were also available for seven trials and were analyzed using the generic inverse variation method.24252728293031 The decline in percent-predicted DLco was not significantly lower in the PH-specific agent group than in the control group (MD, 0.81%; 95% CI, −0.24, 1.87; P = 0.13; I2 = 0%) (Fig. 3C). The mean change from baseline in DLco (mmol·kPa−1·min−1) did not differ significantly between the two groups either (MD, 0.12 mmol·kPa−1·min−1; 95% CI, −0.18, 0.43; P = 0.43; I2 = 0%) (Fig. 3D).
Fig. 3
Pooled effects of PH-specific agents versus controls on changes of lung function; (A) FVC % predicted, (B) FVC L, (C) DLco %, and (D) DLco mmol·kPa−1·min−1.
DLco = diffusion capacity of the lung for carbon monoxide, FVC = forced vital capacity, PH = pulmonary hypertension, MD = mean difference, SD = standard deviation, CI = confidence interval.
Quality of life, exercise capacity, and serious adverse effects
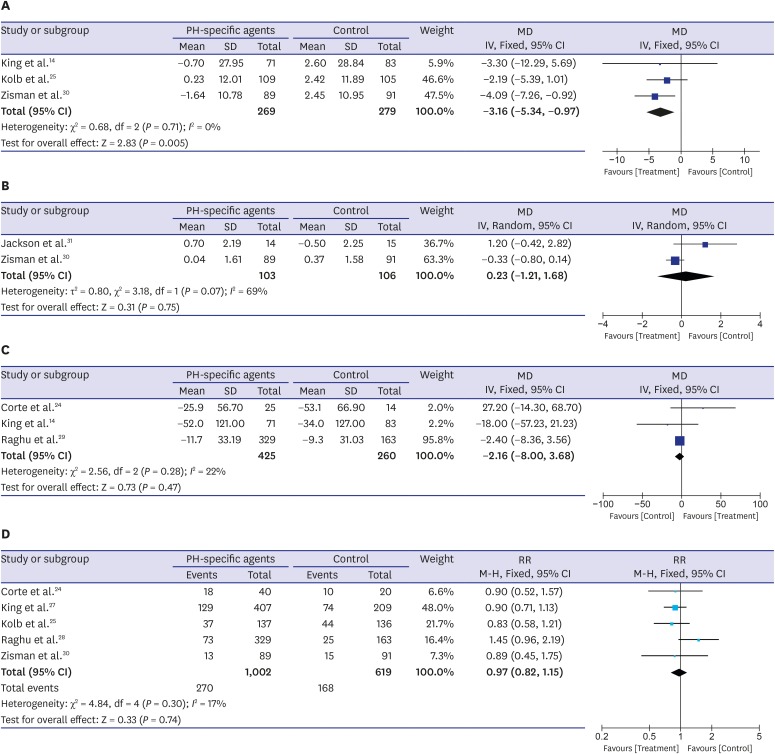
Fig. 4A shows the effectiveness of PH-specific agents in improving the quality of life for IPF patients, measured using the SGRQ total score; these data were reported in three trials.252730 The pooled estimates show a significant improvement in the SGRQ total score in the PH-specific agent group compared with controls (MD, −3.16 points; 95% CI, −5.34, −0.97; P = 0.005; I2 = 0%). However, two RCTs reported data from the Borg dyspnea index score after walk test, and there was no difference between the two groups (MD, 0.23 points; 95% CI, −1.21, 1.68; P = 0.75) (Fig. 4B).3031 Between-trial heterogeneity was substantial (I2 = 69%). Three trials evaluated the effectiveness of PH-specific agents on changing 6MWD results,142428 and no between-group differences were found (MD, −2.16 meter; 95% CI, −8.00, 3.68; P = 0.47; I2 = 22%) (Fig. 4C). Also, as shown in Fig. 4D, serious adverse events were similar between the study and control groups (RR, 0.97; 95% CI, 0.82, 1.15; P = 0.74; I2 = 17%).2425272830
Fig. 4
Pooled effects of PH-specific agents versus controls on changes in clinical parameters and for incidence of serious adverse events; (A) St. George's Respiratory Questionnaire score, (B) Borg dyspnea score after walk test, (C) 6-minute walk distance, and (D) incidence of serious adverse events. A serious adverse event was defined as an event that resulted in death, was life-threatening, resulted in hospitalization or prolongation of hospitalization, resulted in persistent or clinically significant disability or incapacity, was a congenital anomaly or birth defect, or was deemed to be serious for any other reason.
PH = pulmonary hypertension, MD = mean difference, SD = standard deviation, CI = confidence interval.
DISCUSSION
In this systematic review and meta-analysis, we report the following major findings. 1) The use of PH-specific agents did not improve all-cause mortality to end of study among IPF patients. Furthermore, although the included clinical trials showed a moderate degree of heterogeneity in populations and interventions, no clinical benefit in all-cause mortality to end of study was found in any of the sub-group analyses. 2) PH-specific agents yielded small benefits for patient quality of life, as estimated by SGRQ total score. 3) No association was observed between PH-specific agents and secondary outcomes (FVC decline, DLco decline, Borg dyspnea index score, and 6MWD decline). The number of serious adverse events did not differ significantly between the treatment and control groups.
PH is commonly observed to co-occur with IPF, and its presence is associated with a significant negative effect on survival time. In a large registry of 1,344 PH patients, 3-year survival was 44% for patients with PH-associated lung disease.32 Among an examined cohort, patients with established PH and interstitial lung disease had the worst 3-year survival rate of 16%.32 One-year mortality rates were higher in IPF patients with PH awaiting lung transplantation than in those without PH (28% vs. 5.5%, P = 0.002).4 Targeted interventions against PH in IPF patients might be considered a feasible treatment option to improve clinical outcomes. PH-specific agents are regarded as experimental in IPF patients, and the use of these agents is not recommended by current guidelines because of pathophysiologic concerns and the lack of quality data.33334 However, PH-specific agents contribute to vasodilation and remodeling of the pulmonary vasculature, and some studies have reported that they are correlated with better clinical outcomes, including exercise capacity, symptoms, and quality of life.141516 To date, it has not been fully established whether PH-specific agents have a clinical effect on IPF patients.
Mortality to end of study is considered to be the most useful primary endpoint for Phase 3 clinical trials in IPF.35 Mortality-related measures include all-cause mortality, respiratory-related mortality, and IPF-related mortality. Because all-cause mortality during follow-up is the cleanest and most easily interpreted mortality-related endpoint, we selected it as our primary endpoint.35 In this study, we demonstrated that PH-specific agents were not associated with a reduction in all-cause mortality to end of study compared with controls. Although one RCT did report decreased all-cause mortality to end of study, its results were limited by a small sample size and high risk of bias.26 The current findings of our pooled estimates might be explained in the following ways. 1) The development of IPF-associated PH can be explained by hypoxemia-induced vascular remodeling, IPF-specific hyperplasia and fibrosis of the elastic lamina of small pulmonary arteries, in situ thrombosis in small pulmonary arteries, intimal proliferation and fibrosis of the pulmonary venules, and various IPF-mediated cytokine effects.36 Increased pulmonary vascular resistance could result in fixed pulmonary vascular remodeling.37 Thus, PH-specific agents could provoke systemic hypotension if adequate cardiac output is not maintained, and they might not elicit pulmonary vasodilatation.37 2) PH-specific agents could attenuate the physiological hypoxic vasoconstrictor mechanism and increase blood flow to weakly ventilated lung areas. This could eventually lead to concerns about worsening the preexisting ventilation/perfusion mismatch and shunting, causing hypoxia.37
The clinical efficacy of PH-specific agents has been well established in patients with primary PH.34 It also has been proposed that PH-specific agents might benefit IPF patients with confirmed PH.15 However, little available research has focused on the clinical efficacy of PH-specific agents for IPF patients with confirmed PH; we could not conduct a meta-analysis of such studies due to insufficient data. Most of the studies that we excluded from our meta-analysis were non-randomized or were experimental trials focusing on short-term hemodynamic changes from baseline.373839404142 We found three studies that examined treatment outcomes in IPF patients with confirmed PH.152428 The first study examined the effects of ambrisentan on IPF progression and performed stratified analyses of patients with confirmed PH.28 The presence of baseline PH was not associated with a significant difference in IPF progression (HR, 2.42; 95% CI, 0.79, 7.38; P = 0.12 for ambrisentan vs. placebo).28 In the second RCT, which investigated the clinical efficacy of bosentan, no difference was found in invasive pulmonary hemodynamics, functional capacity, or symptoms between the bosentan and placebo groups at endpoint.24 Post hoc analysis revealed that sildenafil treatment resulted in better exercise capacity and quality of life than placebo in IPF patients with right ventricular systolic dysfunction.15
Parameters such as lung function, quality of life, dyspnea degree, and exercise capacity are commonly estimated in clinical trials evaluating the efficacy of treatments for IPF patients.43 Our pooled estimates indicate that PH-specific agents significantly affected quality of life, as measured by SGRQ total score. Additionally, although we identified two trials that examined quality of life, we had to exclude them from our pooled analyses because they used a scoring system other than the SGRQ or because they reported insufficient outcomes.1627 The results of those trials were inconsistent with those of our pooled estimates. A large-scale RCT found no significant difference between the bosentan and control groups with regard to quality of life as measured by the 36-item Short-Form Health Survey and the EuroQol Group Five Dimension Self-Report questionnaire.27 Another exploratory study reported that bosentan treatment had a minimal effect on quality of life after 12 months and had beneficial effects on the SGRQ scores of the subset of IPF patients proven by surgical lung biopsy.16
To the best of our knowledge, this is the first meta-analysis to estimate the clinical impact of PH-specific agents compared with controls. However, our study has several limitations. First, because our meta-analysis was performed on a small number of trials, it might be insufficient to allow generalizations from our findings. Second, our results are based on findings from RCTs that used various PH-specific agents and a heterogeneous patient population. In particular, several studies allowed the use of concomitant medications, including anti-fibrotic agents and immunosuppressive agents.142425262728 Those medications could have influenced our findings. Additionally, we could not find any RCTs that evaluated prostacyclin pathway agonists or sGCSs among the classes of PH-specific agents available; the clinical effects of those agents should be more thoroughly investigated in the future. Third, the study duration ranged from 3 to 24 months of treatment with PH-specific agents, and a study duration of less than 6 months might be insufficient to characterize the clinical efficacy of treatment. Finally, because two trials reported findings extracted from the Sildenafil Trial of Exercise Performance in IPF study, some patients were overlapped in the process of pooling the estimates,2530 which could have elicited selection bias. We included both trials in the present study because they used differently defined populations.
In conclusion, our systematic review and meta-analysis reveals that there is insufficient evidence to suggest a clinical benefit of PH-specific agents among IPF patients, even though these agents provided a small benefit in terms of quality of life improvement. Our findings support the current international guidelines for IPF treatment, which recommend against administering PH-specific agents. Questions remain about whether PH-specific agents offer better outcomes for specific patient subgroups, such as IPF patients with confirmed PH.
 XML Download
XML Download