

 PDF
PDF Citation
Citation Print
Print


Abstract
Cancer immunotherapy, in the form of vaccination, adoptive cellular transfer, or immune checkpoint inhibitors, has emerged as a promising practice within the field of oncology. However, despite the developing field's potential to revolutionize cancer treatment, the presence of immunotherapeutic-resistant tumor cells in many patients present a challenge and limitation to these immunotherapies. These cells not only indicate immunotherapeutic resistance, but also show multi-modal resistance to conventional therapies, abnormal metabolism, stemness, and metastasis. How can immunotherapeutic-resistant tumor cells render multi-malignant phenotypes? We reasoned that the immune-refractory phenotype could be associated with multi-malignant phenotypes and that these phenotypes are linked together by a factor that acts as the master regulator. In this review, we discussed the role of the embryonic transcription factor NANOG as a crucial master regulator we named “common factor” in multi-malignant phenotypes and presented strategies to overcome multi-malignancy in immunotherapeutic-resistant cancer by restraining the NANOG-mediated multi-malignant signaling axis. Strategies that blunt the NANOG axis could improve the clinical management of therapy-refractory cancer.
Go to : 
References
1. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013; 369:122–133.

2. Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, Chow LQ, Vokes EE, Felip E, Holgado E, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015; 373:1627–1639.
3. Ferris RL, Blumenschein G Jr, Fayette J, Guigay J, Colevas AD, Licitra L, Harrington K, Kasper S, Vokes EE, Even C, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. 2016; 375:1856–1867.
4. Yang Y. Cancer immunotherapy: harnessing the immune system to battle cancer. J Clin Invest. 2015; 125:3335–3337.
5. Park YM, Lee SJ, Kim YS, Lee MH, Cha GS, Jung ID, Kang TH, Han HD. Nanoparticle-based vaccine delivery for cancer immunotherapy. Immune Netw. 2013; 13:177–183.
6. Kim JS, Kim YG, Park EJ, Kim B, Lee HK, Hong JT, Kim Y, Han SB. Cell-based immunotherapy for colorectal cancer with cytokine-induced killer cells. Immune Netw. 2016; 16:99–108.
7. Son KJ, Choi KR, Lee SJ, Lee H. Immunogenic cell death induced by ginsenoside rg3: significance in dendritic cell-based anti-tumor immunotherapy. Immune Netw. 2016; 16:75–84.
8. Sanmamed MF, Chen L. A paradigm shift in cancer immunotherapy: from enhancement to normalization. Cell. 2018; 175:313–326.
9. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017; 168:707–723.
10. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014; 515:568–571.
11. Cho JH. Immunotherapy for non-small-cell lung cancer: current status and future obstacles. Immune Netw. 2017; 17:378–391.
12. Noh KH, Lee YH, Jeon JH, Kang TH, Mao CP, Wu TC, Kim TW. Cancer vaccination drives Nanog-dependent evolution of tumor cells toward an immune-resistant and stem-like phenotype. Cancer Res. 2012; 72:1717–1727.
13. Noh KH, Kim BW, Song KH, Cho H, Lee YH, Kim JH, Chung JY, Kim JH, Hewitt SM, Seong SY, et al. Nanog signaling in cancer promotes stem-like phenotype and immune evasion. J Clin Invest. 2012; 122:4077–4093.
14. Song KH, Cho H, Kim S, Lee HJ, Oh SJ, Woo SR, Hong SO, Jang HS, Noh KH, Choi CH, et al. API5 confers cancer stem cell-like properties through the FGF2-NANOG axis. Oncogenesis. 2017; 6:e285.
15. Lee YH, Bae HC, Noh KH, Song KH, Ye SK, Mao CP, Lee KM, Wu TC, Kim TW. Gain of HIF-1α under normoxia in cancer mediates immune adaptation through the AKT/ERK and VEGFA axes. Clin Cancer Res. 2015; 21:1438–1446.
16. Oh SJ, Cho H, Kim S, Noh KH, Song KH, Lee HJ, Woo SR, Kim S, Choi CH, Chung JY, et al. Targeting cyclin d-cdk4/6 sensitizes immune-refractory cancer by blocking the scp3-nanog axis. Cancer Res. 2018; 78:2638–2653.
17. Song KH, Choi CH, Lee HJ, Oh SJ, Woo SR, Hong SO, Noh KH, Cho H, Chung EJ, Kim JH, et al. Hdac1 upregulation by nanog promotes multidrug resistance and a stem-like phenotype in immune edited tumor cells. Cancer Res. 2017; 77:5039–5053.
18. Song KH, Kim JH, Lee YH, Bae HC, Lee HJ, Woo SR, Oh SJ, Lee KM, Yee C, Kim BW, et al. Mitochondrial reprogramming via ATP5H loss promotes multimodal cancer therapy resistance. J Clin Invest. 2018; 128:4098–4114.
19. Jang HS, Woo SR, Song KH, Cho H, Chay DB, Hong SO, Lee HJ, Oh SJ, Chung JY, Kim JH, et al. API5 induces cisplatin resistance through FGFR signaling in human cancer cells. Exp Mol Med. 2017; 49:e374.
20. Woo SR, Lee HJ, Oh SJ, Kim S, Park SH, Lee J, Song KH, Kim TW. Stabilization of HDAC1 via TCL1-pAKT-CHFR axis is a key element for NANOG-mediated multi-resistance and stem-like phenotype in immune-edited tumor cells. Biochem Biophys Res Commun. 2018; 503:1812–1818.
21. Lee HJ, Noh KH, Lee YH, Song KH, Oh SJ, Kim SY, Kim TW. NANOG signaling promotes metastatic capability of immunoedited tumor cells. Clin Exp Metastasis. 2015; 32:429–439.
22. Song KH, Kim SH, Noh KH, Bae HC, Kim JH, Lee HJ, Song J, Kang TH, Kim DW, Oh SJ, et al. Apoptosis inhibitor 5 increases metastasis via erk-mediated mmp expression. BMB Rep. 2015; 48:330–335.
23. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. 2011; 331:1565–1570.
24. O'Donnell JS, Teng MW, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol. 2019; 16:151–167.
25. Schatton T, Frank MH. Antitumor immunity and cancer stem cells. Ann N Y Acad Sci. 2009; 1176:154–169.
26. Frank NY, Schatton T, Frank MH. The therapeutic promise of the cancer stem cell concept. J Clin Invest. 2010; 120:41–50.
27. Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005; 5:275–284.
28. Lytle NK, Barber AG, Reya T. Stem cell fate in cancer growth, progression and therapy resistance. Nat Rev Cancer. 2018; 18:669–680.
29. Wang ML, Chiou SH, Wu CW. Targeting cancer stem cells: emerging role of Nanog transcription factor. Onco Targets Ther. 2013; 6:1207–1220.
30. Bussolati B, Bruno S, Grange C, Ferrando U, Camussi G. Identification of a tumor-initiating stem cell population in human renal carcinomas. FASEB J. 2008; 22:3696–3705.
31. Ezeh UI, Turek PJ, Reijo RA, Clark AT. Human embryonic stem cell genes OCT4, NANOG, STELLAR, and GDF3 are expressed in both seminoma and breast carcinoma. Cancer. 2005; 104:2255–2265.
32. Hart AH, Hartley L, Parker K, Ibrahim M, Looijenga LH, Pauchnik M, Chow CW, Robb L. The pluripotency homeobox gene NANOG is expressed in human germ cell tumors. Cancer. 2005; 104:2092–2098.
33. Hoei-Hansen CE, Almstrup K, Nielsen JE, Brask Sonne S, Graem N, Skakkebaek NE, Leffers H, Rajpert-De Meyts E. Stem cell pluripotency factor NANOG is expressed in human fetal gonocytes, testicular carcinoma in situ and germ cell tumours. Histopathology. 2005; 47:48–56.
34. Lin T, Ding YQ, Li JM. Overexpression of Nanog protein is associated with poor prognosis in gastric adenocarcinoma. Med Oncol. 2012; 29:878–885.
35. Guo Y, Liu S, Wang P, Zhao S, Wang F, Bing L, Zhang Y, Ling EA, Gao J, Hao A. Expression profile of embryonic stem cell-associated genes Oct4, Sox2 and Nanog in human gliomas. Histopathology. 2011; 59:763–775.
36. Wen J, Park JY, Park KH, Chung HW, Bang S, Park SW, Song SY. Oct4 and Nanog expression is associated with early stages of pancreatic carcinogenesis. Pancreas. 2010; 39:622–626.
37. Chiou SH, Yu CC, Huang CY, Lin SC, Liu CJ, Tsai TH, Chou SH, Chien CS, Ku HH, Lo JF. Positive correlations of Oct-4 and Nanog in oral cancer stem-like cells and high-grade oral squamous cell carcinoma. Clin Cancer Res. 2008; 14:4085–4095.
38. Zhou X, Zhou YP, Huang GR, Gong BL, Yang B, Zhang DX, Hu P, Xu SR. Expression of the stem cell marker, Nanog, in human endometrial adenocarcinoma. Int J Gynecol Pathol. 2011; 30:262–270.
39. Nagata T, Shimada Y, Sekine S, Hori R, Matsui K, Okumura T, Sawada S, Fukuoka J, Tsukada K. Prognostic significance of NANOG and KLF4 for breast cancer. Breast Cancer. 2014; 21:96–101.
40. Lee M, Nam EJ, Kim SW, Kim S, Kim JH, Kim YT. Prognostic impact of the cancer stem cell-related marker NANOG in ovarian serous carcinoma. Int J Gynecol Cancer. 2012; 22:1489–1496.
41. Chiou SH, Wang ML, Chou YT, Chen CJ, Hong CF, Hsieh WJ, Chang HT, Chen YS, Lin TW, Hsu HS, et al. Coexpression of Oct4 and Nanog enhances malignancy in lung adenocarcinoma by inducing cancer stem cell-like properties and epithelial-mesenchymal transdifferentiation. Cancer Res. 2010; 70:10433–10444.
42. Han J, Zhang F, Yu M, Zhao P, Ji W, Zhang H, Wu B, Wang Y, Niu R. RNA interference-mediated silencing of NANOG reduces cell proliferation and induces G0/G1 cell cycle arrest in breast cancer cells. Cancer Lett. 2012; 321:80–88.
43. Choi SC, Choi JH, Park CY, Ahn CM, Hong SJ, Lim DS. Nanog regulates molecules involved in stemness and cell cycle-signaling pathway for maintenance of pluripotency of P19 embryonal carcinoma stem cells. J Cell Physiol. 2012; 227:3678–3692.
44. Bourguignon LY, Peyrollier K, Xia W, Gilad E. Hyaluronan-cd44 interaction activates stem cell marker nanog, stat-3-mediated mdr1 gene expression, and ankyrin-regulated multidrug efflux in breast and ovarian tumor cells. J Biol Chem. 2008; 283:17635–17651.
45. Xu F, Dai C, Zhang R, Zhao Y, Peng S, Jia C. Nanog: a potential biomarker for liver metastasis of colorectal cancer. Dig Dis Sci. 2012; 57:2340–2346.
46. Tsai LL, Yu CC, Chang YC, Yu CH, Chou MY. Markedly increased Oct4 and Nanog expression correlates with cisplatin resistance in oral squamous cell carcinoma. J Oral Pathol Med. 2011; 40:621–628.
47. Siu MK, Wong ES, Kong DS, Chan HY, Jiang L, Wong OG, Lam EW, Chan KK, Ngan HY, Le XF, et al. Stem cell transcription factor NANOG controls cell migration and invasion via dysregulation of E-cadherin and FoxJ1 and contributes to adverse clinical outcome in ovarian cancers. Oncogene. 2013; 32:3500–3509.
48. Biswas G, Anandatheerthavarada HK, Avadhani NG. Mechanism of mitochondrial stress-induced resistance to apoptosis in mitochondrial DNA-depleted C2C12 myocytes. Cell Death Differ. 2005; 12:266–278.
49. Fulda S, Galluzzi L, Kroemer G. Targeting mitochondria for cancer therapy. Nat Rev Drug Discov. 2010; 9:447–464.
50. Cuezva JM, Krajewska M, de Heredia ML, Krajewski S, Santamaría G, Kim H, Zapata JM, Marusawa H, Chamorro M, Reed JC. The bioenergetic signature of cancer: a marker of tumor progression. Cancer Res. 2002; 62:6674–6681.
51. Isidoro A, Martínez M, Fernández PL, Ortega AD, Santamaría G, Chamorro M, Reed JC, Cuezva JM. Alteration of the bioenergetic phenotype of mitochondria is a hallmark of breast, gastric, lung and oesophageal cancer. Biochem J. 2004; 378:17–20.
52. Shin YK, Yoo BC, Chang HJ, Jeon E, Hong SH, Jung MS, Lim SJ, Park JG. Down-regulation of mitochondrial F1F0-ATP synthase in human colon cancer cells with induced 5-fluorouracil resistance. Cancer Res. 2005; 65:3162–3170.
53. Liberti MV, Locasale JW. The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci. 2016; 41:211–218.
54. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009; 324:1029–1033.
55. Easwaran H, Tsai HC, Baylin SB. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell. 2014; 54:716–727.
Go to :
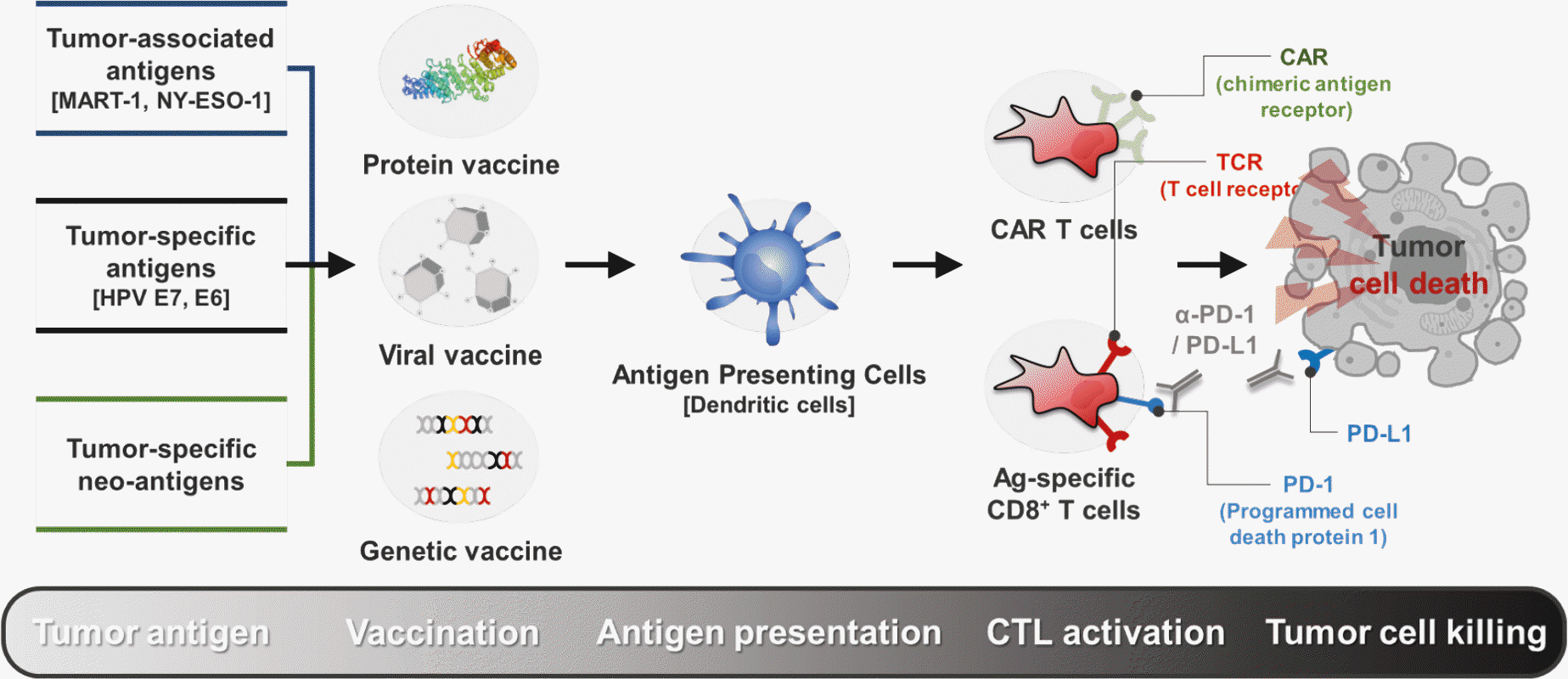 | Figure 1.Emerging trends in the cancer immunotherapy. Cancer immunotherapy induces the patient's immune system's ability to recognize and kill tumor cells through tumor-specific Ags, tumor-associated Ags, and killer T cells. Immunotherapy can be materialized by vaccination or ACT, involving the transfer of CTLs that have been expanded to target tumor Ags. Recently, ICB therapy has taken the center stage in cancer treatment to address the ability of tumors to evade the immune system. MART1, melanoma Ag recognized by T cells 1; NY-ESO-1, cancer/testis Ag 1; HPV, human papillomavirus; CAR, chimeric Ag receptor. |
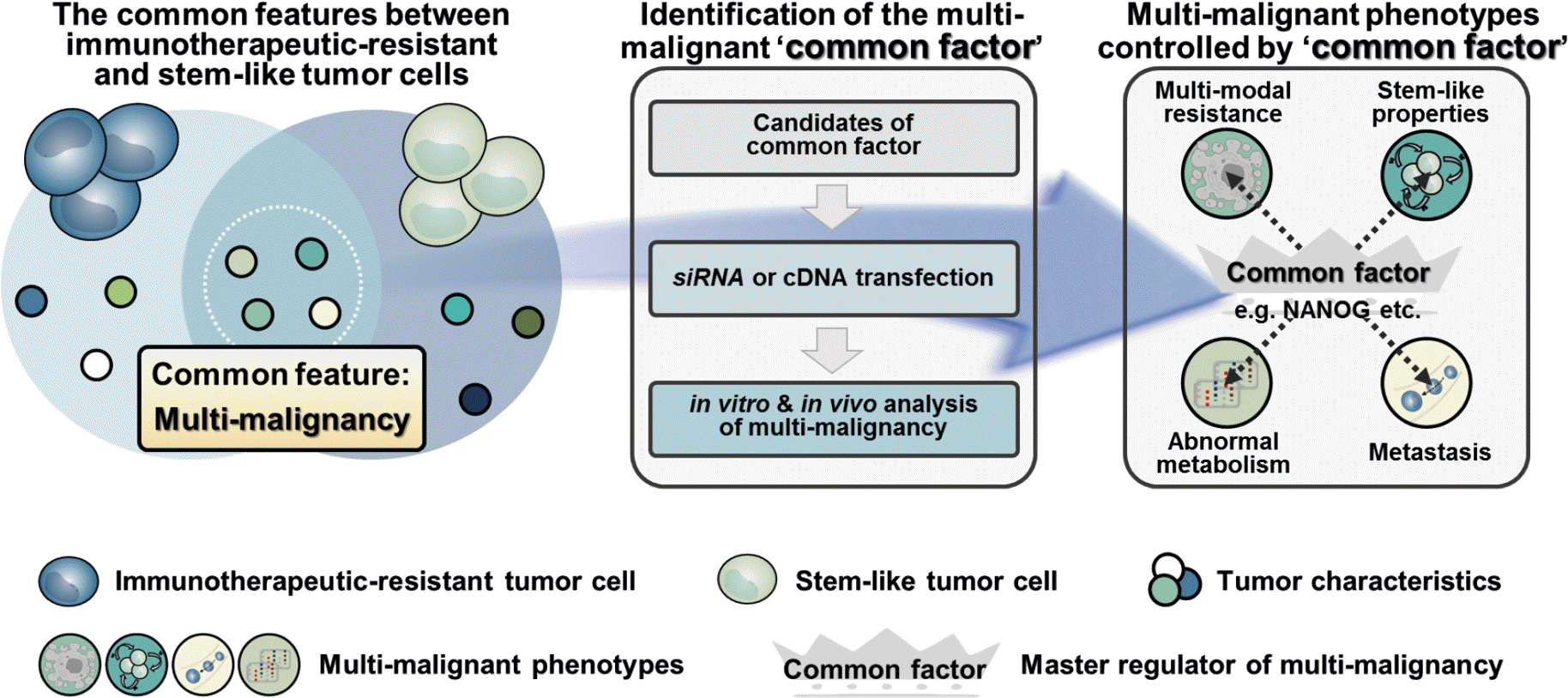 | Figure 2.Concept of common factor which regulates multi-malignant phenotypes. Phenotypes common to stem-like tumor cells and immunotherapeutic-resistant tumor cells provide a first clue to identify a common factor that confers the multi-malignant phenotypes. |
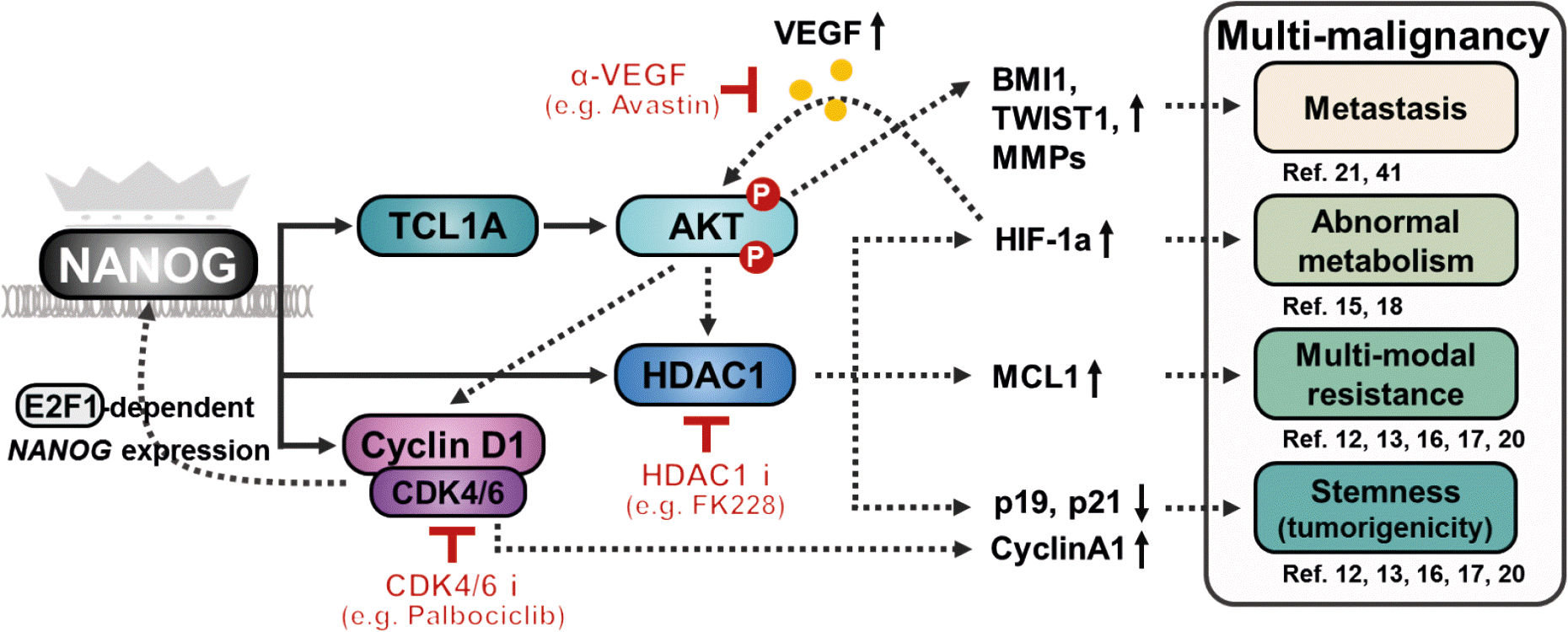 | Figure 3.Key targets for overcoming NANOG-mediated multi-malignancy. Diagrammatic representation of the NANOG signaling axis, which trigger the multi-malignant properties of tumor cells, and different therapeutic strategies to target the NANOG axis. TWIST1, twist-related protein 1; MMP, matrix metallopeptidase; HIF-1, hypoxia-inducible factor-1; MCL1, myeloid cell leukemia 1; BMI1, B lymphoma Mo-MLV insertion region 1 homolog. |
 XML Download
XML Download