

 PDF
PDF Citation
Citation Print
Print


Abstract
Tregs have a role in immunological tolerance and immune homeostasis by suppressing immune reactions, and its therapeutic potential is critical in autoimmune diseases and cancers. There have been multiple studies conducted on Tregs because of their roles in immune suppression and therapeutic potential. In tumor immunity, Tregs can promote the development and progression of tumors by preventing effective anti-tumor immune responses in tumor-bearing hosts. High infiltration of Tregs into tumor tissue results in poor survival in various types of cancer patients. Identifying factors specifically expressed in Tregs that affect the maintenance of stability and function of Tregs is important for understanding cancer pathogenesis and identifying therapeutic targets. Thus, manipulation of Tregs is a promising anticancer strategy, but finding markers for Treg-specific depletion and controlling these cells require fine-tuning and further research. Here, we discuss the role of Tregs in cancer and the development of Treg-targeted therapies to promote cancer immunotherapy.
Go to : 
References
1. Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. 2012; 30:531–564.

2. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+ CD25+ regulatory T cells. Nat Immunol. 2003; 4:330–336.
3. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003; 299:1057–1061.
4. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, Kelly TE, Saulsbury FT, Chance PF, Ochs HD. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001; 27:20–21.
5. Motz GT, Coukos G. Deciphering and reversing tumor immune suppression. Immunity. 2013; 39:61–73.
6. Baecher-Allan C, Brown JA, Freeman GJ, Hafler DA. CD4+ CD25 high regulatory cells in human peripheral blood. J Immunol. 2001; 167:1245–1253.
7. Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, Gajewski TF. Up-regulation of PD-L1, IDO, and Tregs in the melanoma tumor microenvironment is driven by CD8+ T cells. Sci Transl Med. 2013; 5:200ra116.
8. Williams JB, Horton BL, Zheng Y, Duan Y, Powell JD, Gajewski TF. The EGR2 targets LAG-3 and 4–1BB describe and regulate dysfunctional antigen-specific CD8+ T cells in the tumor microenvironment. J Exp Med. 2017; 214:381–400.
9. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012; 12:252–264.
10. Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016; 8:328rv4.
11. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012; 366:2455–2465.
12. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010; 363:711–723.
13. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012; 366:2443–2454.
14. Togashi Y, Nishikawa H. Regulatory T cells: molecular and cellular basis for immunoregulation. Curr Top Microbiol Immunol. 2017; 410:3–27.
15. Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol. 2010; 10:490–500.
16. Hsieh CS, Zheng Y, Liang Y, Fontenot JD, Rudensky AY. An intersection between the self-reactive regulatory and nonregulatory T cell receptor repertoires. Nat Immunol. 2006; 7:401–410.
17. Wong J, Obst R, Correia-Neves M, Losyev G, Mathis D, Benoist C. Adaptation of TCR repertoires to self-peptides in regulatory and nonregulatory CD4+ T cells. J Immunol. 2007; 178:7032–7041.
18. Yadav M, Stephan S, Bluestone JA. Peripherally induced Tregs – role in immune homeostasis and autoimmunity. Front Immunol. 2013; 4:232.
19. Ziegler SF. FOXP3: of mice and men. Annu Rev Immunol. 2006; 24:209–226.
20. Roychoudhuri R, Hirahara K, Mousavi K, Clever D, Klebanoff CA, Bonelli M, Sciumè G, Zare H, Vahedi G, Dema B, et al. BACH2 represses effector programs to stabilize Treg-mediated immune homeostasis. Nature. 2013; 498:506–510.
21. Igarashi K, Kurosaki T, Roychoudhuri R. BACH transcription factors in innate and adaptive immunity. Nat Rev Immunol. 2017; 17:437–450.
22. Kim EH, Gasper DJ, Lee SH, Plisch EH, Svaren J, Suresh M. Bach2 regulates homeostasis of Foxp3+ regulatory T cells and protects against fatal lung disease in mice. J Immunol. 2014; 192:985–995.
23. Overacre-Delgoffe AE, Chikina M, Dadey RE, Yano H, Brunazzi EA, Shayan G, Horne W, Moskovitz JM, Kolls JK, Sander C, et al. Interferon-γ drives Treg fragility to promote anti-tumor immunity. Cell. 2017; 169:1130–1141. e11.
24. Sarris M, Andersen KG, Randow F, Mayr L, Betz AG. Neuropilin-1 expression on regulatory T cells enhances their interactions with dendritic cells during antigen recognition. Immunity. 2008; 28:402–413.
25. Getnet D, Grosso JF, Goldberg MV, Harris TJ, Yen HR, Bruno TC, Durham NM, Hipkiss EL, Pyle KJ, Wada S. A role for the transcription factor Helios in human CD4+ CD25+ regulatory T cells. Mol Immunol. 2010; 47:1595–1600.
26. Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature. 2010; 463:808–812.
27. Lathrop SK, Santacruz NA, Pham D, Luo J, Hsieh CS. Antigen-specific peripheral shaping of the natural regulatory T cell population. J Exp Med. 2008; 205:3105–3117.
28. Tran DQ, Ramsey H, Shevach EM. Induction of FOXP3 expression in naive human CD4+ FOXP3 T cells by T-cell receptor stimulation is transforming growth factor-beta dependent but does not confer a regulatory phenotype. Blood. 2007; 110:2983–2990.
29. Liu W, Putnam AL, Xu-Yu Z, Szot GL, Lee MR, Zhu S, Gottlieb PA, Kapranov P, Gingeras TR, Fazekas de St Groth B, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med. 2006; 203:1701–1711.
30. Seddiki N, Santner-Nanan B, Martinson J, Zaunders J, Sasson S, Landay A, Solomon M, Selby W, Alexander SI, Nanan R, et al. Expression of interleukin (IL)-2 and IL-7 receptors discriminates between human regulatory and activated T cells. J Exp Med. 2006; 203:1693–1700.
31. Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, Parizot C, Taflin C, Heike T, Valeyre D, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity. 2009; 30:899–911.
32. Thornton AM, Shevach EM. CD4+ CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J Exp Med. 1998; 188:287–296.
33. Takahashi T, Kuniyasu Y, Toda M, Sakaguchi N, Itoh M, Iwata M, Shimizu J, Sakaguchi S. Immunologic self-tolerance maintained by CD25+ CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. Int Immunol. 1998; 10:1969–1980.
34. Steinbrink K, Wölfl M, Jonuleit H, Knop J, Enk AH. Induction of tolerance by IL-10-treated dendritic cells. J Immunol. 1997; 159:4772–4780.
35. Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, Cross R, Sehy D, Blumberg RS, Vignali DA. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. 2007; 450:566–569.
36. Turnis ME, Sawant DV, Szymczak-Workman AL, Andrews LP, Delgoffe GM, Yano H, Beres AJ, Vogel P, Workman CJ, Vignali DA. Interleukin-35 limits anti-tumor immunity. Immunity. 2016; 44:316–329.
37. Jarnicki AG, Lysaght J, Todryk S, Mills KH. Suppression of antitumor immunity by IL-10 and TGF-beta-producing T cells infiltrating the growing tumor: influence of tumor environment on the induction of CD4+ and CD8+ regulatory T cells. J Immunol. 2006; 177:896–904.
38. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, Chen JF, Enjyoji K, Linden J, Oukka M, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007; 204:1257–1265.
39. Wilson JM, Ross WG, Agbai ON, Frazier R, Figler RA, Rieger J, Linden J, Ernst PB. The A2B adenosine receptor impairs the maturation and immunogenicity of dendritic cells. J Immunol. 2009; 182:4616–4623.
40. Uyttenhove C, Pilotte L, Théate I, Stroobant V, Colau D, Parmentier N, Boon T, Van den Eynde BJ. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003; 9:1269–1274.
41. Saleh R, Elkord E. Treg-mediated acquired resistance to immune checkpoint inhibitors. Cancer Lett. 2019; 457:168–179.
42. Burchell JT, Strickland DH, Stumbles PA. The role of dendritic cells and regulatory T cells in the regulation of allergic asthma. Pharmacol Ther. 2010; 125:1–10.
43. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, Nomura T, Sakaguchi S. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008; 322:271–275.
44. Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, Bulashevska A, Petersen BS, Schäffer AA, Grüning BA, et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med. 2014; 20:1410–1416.
45. Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, Schickel JN, Tran DQ, Stoddard J, Zhang Y, et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science. 2014; 345:1623–1627.
46. Perez VL, Van Parijs L, Biuckians A, Zheng XX, Strom TB, Abbas AK. Induction of peripheral T cell tolerance in vivo requires CTLA-4 engagement. Immunity. 1997; 6:411–417.
47. Onizuka S, Tawara I, Shimizu J, Sakaguchi S, Fujita T, Nakayama E. Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor alpha) monoclonal antibody. Cancer Res. 1999; 59:3128–3133.
48. Shimizu J, Yamazaki S, Sakaguchi S. Induction of tumor immunity by removing CD25+ CD4+ T cells: a common basis between tumor immunity and autoimmunity. J Immunol. 1999; 163:5211–5218.
49. Tada Y, Togashi Y, Kotani D, Kuwata T, Sato E, Kawazoe A, Doi T, Wada H, Nishikawa H, Shitara K. Targeting VEGFR2 with Ramucirumab strongly impacts effector/activated regulatory T cells and CD8+ T cells in the tumor microenvironment. J Immunother Cancer. 2018; 6:106.
50. Saito T, Nishikawa H, Wada H, Nagano Y, Sugiyama D, Atarashi K, Maeda Y, Hamaguchi M, Ohkura N, Sato E, et al. Two FOXP3+ CD4+ T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat Med. 2016; 22:679–684.
51. Akimova T, Zhang T, Negorev D, Singhal S, Stadanlick J, Rao A, Annunziata M, Levine MH, Beier UH, Diamond JM, et al. Human lung tumor FOXP3+ Tregs upregulate four “Treg-locking” transcription factors. JCI Insight. 2017; 2:94075.
52. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004; 10:942–949.
53. Takeuchi Y, Nishikawa H. Roles of regulatory T cells in cancer immunity. Int Immunol. 2016; 28:401–409.
54. Gobert M, Treilleux I, Bendriss-Vermare N, Bachelot T, Goddard-Leon S, Arfi V, Biota C, Doffin AC, Durand I, Olive D, et al. Regulatory T cells recruited through CCL22/CCR4 are selectively activated in lymphoid infiltrates surrounding primary breast tumors and lead to an adverse clinical outcome. Cancer Res. 2009; 69:2000–2009.
55. Tan MC, Goedegebuure PS, Belt BA, Flaherty B, Sankpal N, Gillanders WE, Eberlein TJ, Hsieh CS, Linehan DC. Disruption of CCR5-dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J Immunol. 2009; 182:1746–1755.
56. Kryczek I, Wei S, Zhu G, Myers L, Mottram P, Cheng P, Chen L, Coukos G, Zou W. Relationship between B7-H4, regulatory T cells, and patient outcome in human ovarian carcinoma. Cancer Res. 2007; 67:8900–8905.
57. Ladoire S, Martin F, Ghiringhelli F. Prognostic role of FOXP3+ regulatory T cells infiltrating human carcinomas: the paradox of colorectal cancer. Cancer Immunol Immunother. 2011; 60:909–918.
58. Salama P, Phillips M, Grieu F, Morris M, Zeps N, Joseph D, Platell C, Iacopetta B. Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. J Clin Oncol. 2009; 27:186–192.
59. Badoual C, Hans S, Rodriguez J, Peyrard S, Klein C, Agueznay NH, Mosseri V, Laccourreye O, Bruneval P, Fridman WH, et al. Prognostic value of tumor-infiltrating CD4+ T-cell subpopulations in head and neck cancers. Clin Cancer Res. 2006; 12:465–472.
60. Petersen RP, Campa MJ, Sperlazza J, Conlon D, Joshi MB, Harpole DH Jr, Patz EF Jr. Tumor infiltrating Foxp3+ regulatory T-cells are associated with recurrence in pathologic stage I NSCLC patients. Cancer. 2006; 107:2866–2872.
61. Marshall EA, Ng KW, Kung SH, Conway EM, Martinez VD, Halvorsen EC, Rowbotham DA, Vucic EA, Plumb AW, Becker-Santos DD, et al. Emerging roles of T helper 17 and regulatory T cells in lung cancer progression and metastasis. Mol Cancer. 2016; 15:67.
62. Fridman WH, Pagès F, Sautès-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012; 12:298–306.
63. Sugiyama D, Nishikawa H, Maeda Y, Nishioka M, Tanemura A, Katayama I, Ezoe S, Kanakura Y, Sato E, Fukumori Y, et al. Anti-CCR4 mAb selectively depletes effector-type FoxP3+ CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc Natl Acad Sci U S A. 2013; 110:17945–17950.
64. De Simone M, Arrigoni A, Rossetti G, Gruarin P, Ranzani V, Politano C, Bonnal RJ, Provasi E, Sarnicola ML, Panzeri I, et al. Transcriptional landscape of human tissue lymphocytes unveils uniqueness of tumor-infiltrating T regulatory cells. Immunity. 2016; 45:1135–1147.
65. Nishikawa H, Kato T, Tawara I, Saito K, Ikeda H, Kuribayashi K, Allen PM, Schreiber RD, Sakaguchi S, Old LJ, et al. Definition of target antigens for naturally occurring CD4+ CD25+ regulatory T cells. J Exp Med. 2005; 201:681–686.
66. Nishikawa H, Kato T, Tawara I, Ikeda H, Kuribayashi K, Allen PM, Schreiber RD, Old LJ, Shiku H. IFN-gamma controls the generation/activation of CD4+ CD25+ regulatory T cells in antitumor immune response. J Immunol. 2005; 175:4433–4440.
67. Pace L, Tempez A, Arnold-Schrauf C, Lemaitre F, Bousso P, Fetler L, Sparwasser T, Amigorena S. Regulatory T cells increase the avidity of primary CD8+ T cell responses and promote memory. Science. 2012; 338:532–536.
68. Maeda Y, Nishikawa H, Sugiyama D, Ha D, Hamaguchi M, Saito T, Nishioka M, Wing JB, Adeegbe D, Katayama I, et al. Detection of self-reactive CD8+ T cells with an anergic phenotype in healthy individuals. Science. 2014; 346:1536–1540.
69. Nishikawa H, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Curr Opin Immunol. 2014; 27:1–7.
70. Kurose K, Ohue Y, Wada H, Iida S, Ishida T, Kojima T, Doi T, Suzuki S, Isobe M, Funakoshi T, et al. Phase Ia study of FoxP3+ CD4 Treg depletion by infusion of a humanized anti-CCR4 antibody, KW-0761, in cancer patients. Clin Cancer Res. 2015; 21:4327–4336.
71. Jia Z, Zhao R, Tian Y, Huang Z, Tian Z, Shen Z, Wang Q, Wang J, Fu X, Wu Y, et al. A novel splice variant of FR4 predominantly expressed in CD4+ CD25+ regulatory T cells. Immunol Invest. 2009; 38:718–729.
72. Miyara M, Chader D, Sage E, Sugiyama D, Nishikawa H, Bouvry D, Claër L, Hingorani R, Balderas R, Rohrer J, et al. Sialyl Lewis x (CD15s) identifies highly differentiated and most suppressive FOXP3 high regulatory T cells in humans. Proc Natl Acad Sci U S A. 2015; 112:7225–7230.
73. Foss F. Clinical experience with denileukin diftitox (ONTAK). Semin Oncol. 2006; 33:S11–S16.
74. Steitz J, Brück J, Lenz J, Knop J, Tüting T. Depletion of CD25+ CD4+ T cells and treatment with tyrosinase-related protein 2-transduced dendritic cells enhance the interferon alpha-induced, CD8+ T-cell-dependent immune defense of B16 melanoma. Cancer Res. 2001; 61:8643–8646.
75. Rech AJ, Mick R, Martin S, Recio A, Aqui NA, Powell DJ Jr, Colligon TA, Trosko JA, Leinbach LI, Pletcher CH, et al. CD25 blockade depletes and selectively reprograms regulatory T cells in concert with immunotherapy in cancer patients. Sci Transl Med. 2012; 4:134ra62.
76. Jacobs JF, Punt CJ, Lesterhuis WJ, Sutmuller RP, Brouwer HM, Scharenborg NM, Klasen IS, Hilbrands LB, Figdor CG, de Vries IJ, et al. Dendritic cell vaccination in combination with anti-CD25 monoclonal antibody treatment: a phase I/II study in metastatic melanoma patients. Clin Cancer Res. 2010; 16:5067–5078.
77. Romano E, Kusio-Kobialka M, Foukas PG, Baumgaertner P, Meyer C, Ballabeni P, Michielin O, Weide B, Romero P, Speiser DE. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc Natl Acad Sci U S A. 2015; 112:6140–6145.
78. Ribas A. Tumor immunotherapy directed at PD-1. N Engl J Med. 2012; 366:2517–2519.
79. Arce Vargas F, Furness AJ, Litchfield K, Joshi K, Rosenthal R, Ghorani E, Solomon I, Lesko MH, Ruef N, Roddie C, et al. Fc effector function contributes to the activity of human anti-CTLA-4 antibodies. Cancer Cell. 2018; 33:649–663. e4.
80. Bulliard Y, Jolicoeur R, Windman M, Rue SM, Ettenberg S, Knee DA, Wilson NS, Dranoff G, Brogdon JL. Activating Fc γ receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J Exp Med. 2013; 210:1685–1693.
81. Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, Korman AJ. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res. 2013; 1:32–42.
82. Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, Roddie C, Henry JY, Yagita H, Wolchok JD, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med. 2013; 210:1695–1710.
83. van Olffen RW, Koning N, van Gisbergen KP, Wensveen FM, Hoek RM, Boon L, Hamann J, van Lier RA, Nolte MA. GITR triggering induces expansion of both effector and regulatory CD4+ T cells in vivo. J Immunol. 2009; 182:7490–7500.
84. Nishikawa H, Kato T, Hirayama M, Orito Y, Sato E, Harada N, Gnjatic S, Old LJ, Shiku H. Regulatory T cell-resistant CD8+ T cells induced by glucocorticoid-induced tumor necrosis factor receptor signaling. Cancer Res. 2008; 68:5948–5954.
85. Buchan SL, Rogel A, Al-Shamkhani A. The immunobiology of CD27 and OX40 and their potential as targets for cancer immunotherapy. Blood. 2018; 131:39–48.
86. Curti BD, Kovacsovics-Bankowski M, Morris N, Walker E, Chisholm L, Floyd K, Walker J, Gonzalez I, Meeuwsen T, Fox BA, et al. OX40 is a potent immune-stimulating target in late-stage cancer patients. Cancer Res. 2013; 73:7189–7198.
87. Herman AE, Freeman GJ, Mathis D, Benoist C. CD4+ CD25+ T regulatory cells dependent on ICOS promote regulation of effector cells in the prediabetic lesion. J Exp Med. 2004; 199:1479–1489.
88. Burmeister Y, Lischke T, Dahler AC, Mages HW, Lam KP, Coyle AJ, Kroczek RA, Hutloff A. ICOS controls the pool size of effector-memory and regulatory T cells. J Immunol. 2008; 180:774–782.
89. Nagase H, Takeoka T, Urakawa S, Morimoto-Okazawa A, Kawashima A, Iwahori K, Takiguchi S, Nishikawa H, Sato E, Sakaguchi S, et al. ICOS+ Foxp3+ TILs in gastric cancer are prognostic markers and effector regulatory T cells associated with Helicobacter pylori. Int J Cancer. 2017; 140:686–695.
90. Burris HA, Callahan MK, Tolcher AW, Kummar S, Falchook GS, Pachynski RK, Tykodi SS, Gibney GT, Seiwert TY, Gainor JF, et al. Phase 1 safety of ICOS agonist antibody JTX-2011 alone and with nivolumab (nivo) in advanced solid tumors; predicted vs observed pharmacokinetics (PK) in ICONIC. J Clin Oncol. 2017; 35:3033.
91. Kurtulus S, Sakuishi K, Ngiow SF, Joller N, Tan DJ, Teng MW, Smyth MJ, Kuchroo VK, Anderson AC. TIGIT predominantly regulates the immune response via regulatory T cells. J Clin Invest. 2015; 125:4053–4062.
92. Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C, Xia J, Tan TG, Sefik E, Yajnik V, et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity. 2014; 40:569–581.
93. Johnston RJ, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y, Park S, Javinal V, Chiu H, Irving B, et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8+ T cell effector function. Cancer Cell. 2014; 26:923–937.
94. Scurr M, Ladell K, Besneux M, Christian A, Hockey T, Smart K, Bridgeman H, Hargest R, Phillips S, Davies M, et al. Highly prevalent colorectal cancer-infiltrating LAP+ Foxp3− T cells exhibit more potent immunosuppressive activity than Foxp3+ regulatory T cells. Mucosal Immunol. 2014; 7:428–439.
95. Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity. 2016; 44:989–1004.
96. Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, Zheng XX, Strom TB, Kuchroo VK. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. 2005; 6:1245–1252.
97. Das M, Zhu C, Kuchroo VK. Tim-3 and its role in regulating anti-tumor immunity. Immunol Rev. 2017; 276:97–111.
98. Sakuishi K, Ngiow SF, Sullivan JM, Teng MW, Kuchroo VK, Smyth MJ, Anderson AC. TIM3+ FOXP3+ regulatory T cells are tissue-specific promoters of T-cell dysfunction in cancer. OncoImmunology. 2013; 2:e23849.
99. Campbell DJ, Koch MA. Phenotypical and functional specialization of FOXP3+ regulatory T cells. Nat Rev Immunol. 2011; 11:119–130.
100. Ishida T, Ueda R. CCR4 as a novel molecular target for immunotherapy of cancer. Cancer Sci. 2006; 97:1139–1146.
101. Nishikawa H, Sakaguchi S. Regulatory T cells in tumor immunity. Int J Cancer. 2010; 127:759–767.
102. Kurose K, Ohue Y, Oka M. Anti-CCR4 mAb and regulatory T cells. Gan To Kagaku Ryoho. 2013; 40:1150–1155.
103. Zhang B, Chikuma S, Hori S, Fagarasan S, Honjo T. Nonoverlapping roles of PD-1 and FoxP3 in maintaining immune tolerance in a novel autoimmune pancreatitis mouse model. Proc Natl Acad Sci U S A. 2016; 113:8490–8495.
104. Gianchecchi E, Fierabracci A. Inhibitory receptors and pathways of lymphocytes: the role of PD-1 in Treg development and their involvement in autoimmunity onset and cancer progression. Front Immunol. 2018; 9:2374.
105. Kamada T, Togashi Y, Tay C, Ha D, Sasaki A, Nakamura Y, Sato E, Fukuoka S, Tada Y, Tanaka A, et al. PD-1+ regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc Natl Acad Sci U S A. 2019; 116:9999–10008.
106. Colak S, Ten Dijke P. Targeting TGF-β signaling in cancer. Trends Cancer. 2017; 3:56–71.
107. Holmgaard RB, Schaer DA, Li Y, Castaneda SP, Murphy MY, Xu X, Inigo I, Dobkin J, Manro JR, Iversen PW, et al. Targeting the TGFβ pathway with galunisertib, a TGFβ RI small molecule inhibitor, promotes anti-tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint blockade. J Immunother Cancer. 2018; 6:47.
108. Strauss J, Heery CR, Schlom J, Madan RA, Cao L, Kang Z, Lamping E, Marté JL, Donahue RN, Grenga I, et al. Phase I trial of M7824 (MSB0011359C), a bifunctional fusion protein targeting PD-L1 and TGFβ, in advanced solid tumors. Clin Cancer Res. 2018; 24:1287–1295.
109. Ahmad S, Abu-Eid R, Shrimali R, Webb M, Verma V, Doroodchi A, Berrong Z, Samara R, Rodriguez PC, Mkrtichyan M, et al. Differential PI3Kδ signaling in CD4+ T-cell subsets enables selective targeting of T regulatory cells to enhance cancer immunotherapy. Cancer Res. 2017; 77:1892–1904.
110. Ali K, Soond DR, Pineiro R, Hagemann T, Pearce W, Lim EL, Bouabe H, Scudamore CL, Hancox T, Maecker H, et al. Inactivation of PI(3)K p110δ breaks regulatory T-cell-mediated immune tolerance to cancer. Nature. 2014; 510:407–411.
111. Huynh A, DuPage M, Priyadharshini B, Sage PT, Quiros J, Borges CM, Townamchai N, Gerriets VA, Rathmell JC, Sharpe AH, et al. Control of PI(3) kinase in Treg cells maintains homeostasis and lineage stability. Nat Immunol. 2015; 16:188–196.
112. Shrestha S, Yang K, Guy C, Vogel P, Neale G, Chi H. Treg cells require the phosphatase PTEN to restrain T H1 and T FH cell responses. Nat Immunol. 2015; 16:178–187.
113. Imagawa J, Tanaka H, Okada M, Nakamae H, Hino M, Murai K, Ishida Y, Kumagai T, Sato S, Ohashi K, et al. Discontinuation of dasatinib in patients with chronic myeloid leukaemia who have maintained deep molecular response for longer than 1 year (DADI trial): a multicentre phase 2 trial. Lancet Haematol. 2015; 2:e528–e535.
114. Vahl JC, Drees C, Heger K, Heink S, Fischer JC, Nedjic J, Ohkura N, Morikawa H, Poeck H, Schallenberg S, et al. Continuous T cell receptor signals maintain a functional regulatory T cell pool. Immunity. 2014; 41:722–736.
115. Ohta A, Sitkovsky M. Extracellular adenosine-mediated modulation of regulatory T cells. Front Immunol. 2014; 5:304.
116. Terme M, Pernot S, Marcheteau E, Sandoval F, Benhamouda N, Colussi O, Dubreuil O, Carpentier AF, Tartour E, Taieb J. VEGFA-VEGFR pathway blockade inhibits tumor-induced regulatory T-cell proliferation in colorectal cancer. Cancer Res. 2013; 73:539–549.
117. Zhu P, Hu C, Hui K, Jiang X. The role and significance of VEGFR2+ regulatory T cells in tumor immunity. Onco Targets Ther. 2017; 10:4315–4319.
118. Roland CL, Lynn KD, Toombs JE, Dineen SP, Udugamasooriya DG, Brekken RA. Cytokine levels correlate with immune cell infiltration after anti-VEGF therapy in preclinical mouse models of breast cancer. PLoS One. 2009; 4:e7669.
119. Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet AL, Latreche S, Bergaya S, Benhamouda N, Tanchot C, et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med. 2015; 212:139–148.
Go to :
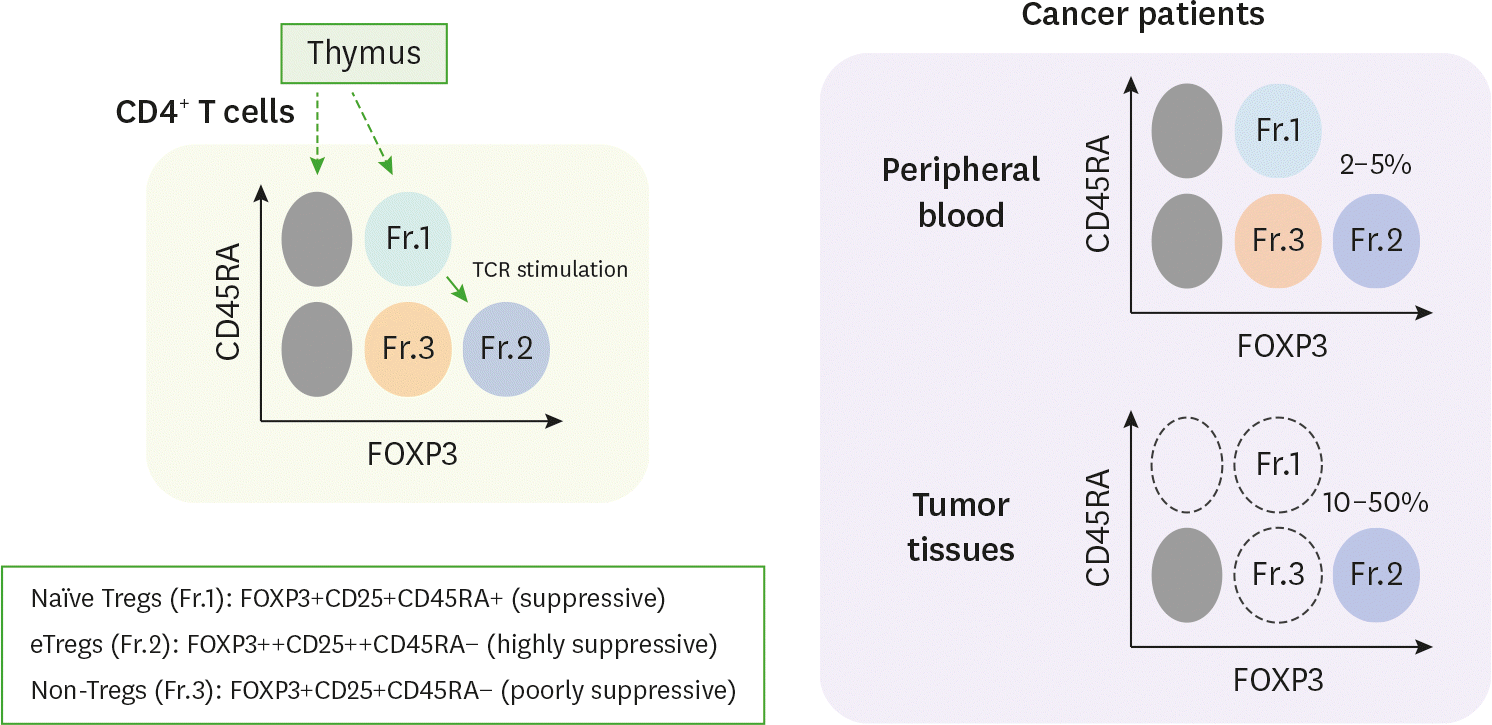 | Figure 1.Classification of human CD4+ FOXP3+ T cells. In humans, CD4+ FOXP3+ T cells can be classified into three subsets: naïve Tregs (Fr.1), eTregs (Fr.2), and non-Tregs (Fr.3). These three fractions can be distinguished based on the expression of CD45RA, cell surface markers of naive T cells, and the transcription factor FOXP3. Moreover, these subpopulations are functionally different in terms of their suppressive activity. Effector Tregs harbor strong immune suppressive activity, but non-Tregs do not possess immune suppressive activity. In the majority of cancer, eTregs predominantly infiltrate into tumor tissues. In general, the frequency of eTregs in cancer patients is 2∼5% in peripheral blood but approximately 10∼50% in the tumor tissues. In contrast, naïve Tregs and FOXP3+ non-Tregs are insufficient or absent altogether. |
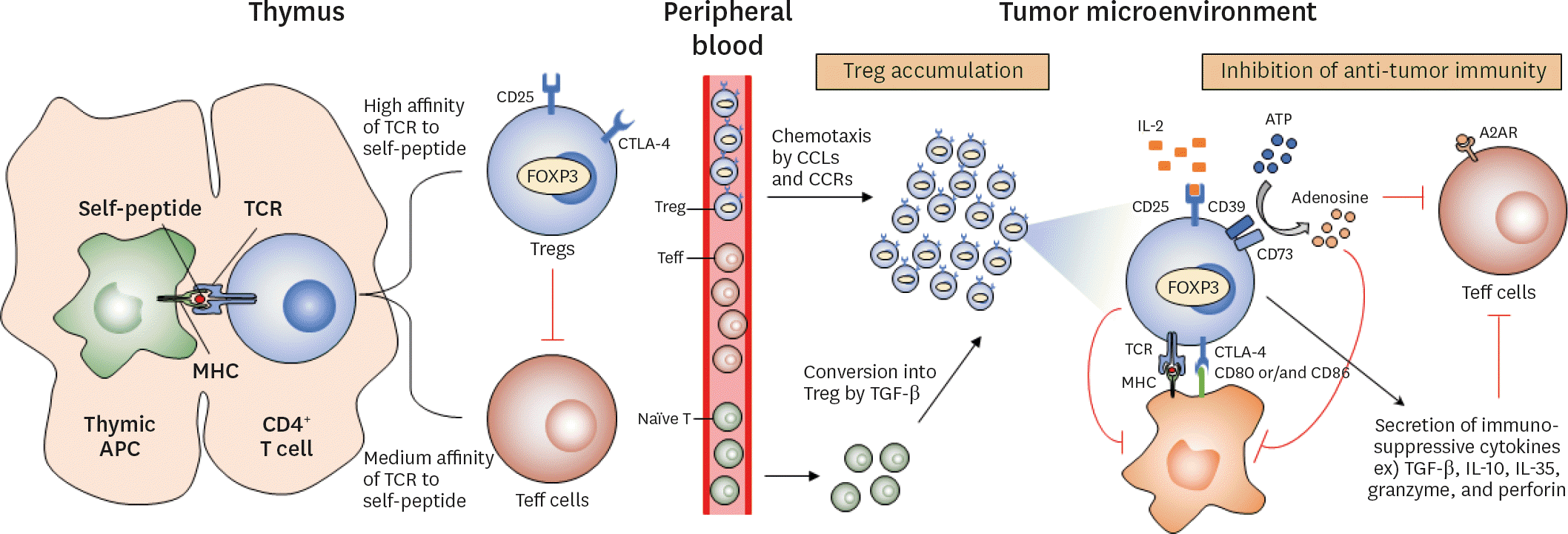 | Figure 2.Role of Tregs in immune-evasion of cancer after differentiation from the thymus. Natural Tregs, generated in the thymus, are initially differentiated from the thymocytes by using thymic “positive selection” based on the binding affinity of TCR to the self- peptides-MHC complexes expressed on thymic APCs. The CD4+ T cells which bind to self-peptide-MHC complexes with the highest affinity are removed through apoptosis, and those that cannot bind at all with the complexes will also be removed because of the absence of TCR stimulation. After strong TCR stimulation, these immature precursor cells undergo IL-2-mediated signaling, thus expressing the master transcription factor FOXP3, which orchestrates the differentiation of these cells into Tregs. By contrast, immature T cells with lower affinity for self-peptide–MHC complexes are also positively selected but differentiate into Teff cells. Even though some Teff cells are auto-reactive, Tregs can block the autoimmunity of Teff cells owing to their higher affinity. These immune cells that have departed from the thymus travel through the blood vessels and move wherever they are needed. In the tumor microenvironment, especially, Tregs expressing the chemokine receptors, such as CCR4, CCR5, CCR8, and CCR10, are recruited to and around the tumors by binding to chemokines including CCL1, CCL5, CCL22, and CCL28 that are secreted from various kinds of tumors. Moreover, Tregs constitutively express the IL-2 receptor subunit-α (also known as CD25) that binds to IL-2 with higher affinity, resulting in the depletion of IL-2 from their surroundings. This leads to the reduction of the availability of this cytokine to Teff cells. Tregs also constitutively express CTLA-4, a checkpoint protein suppressing the immune response, which binds to CD80 and CD86 on APC, thereby transmitting suppressive signals to Teff cells. In addition, Tregs secrete cytokines, such as IL-10, IL-35, and TGF-β, which can decrease the activity of APCs and Teff cells and secrete granzymes and perforins that can directly kill these cells. Moreover, abundant adenosine is produced by Tregs via nucleotidase activity of CD39 and CD73, which provides immunosuppressive signals to Teff cells and APCs through the engagement of adenosine A2AR. |
Table 1.
Ab-drug development status of Treg-targeting therapy
 XML Download
XML Download