

 PDF
PDF Citation
Citation Print
Print


Abstract
Immune checkpoint inhibitors (ICIs), including anti-PD-1 and anti-CTLA-4 therapeutic agents, are now approved by the Food and Drug Administration for treatment of various types of cancer. However, the therapeutic efficacy of ICIs varies among patients and cancer types. Moreover, most patients do not develop durable antitumor responses after ICI therapy due to an ephemeral reversal of T-cell dysfunction. As co-stimulatory receptors play key roles in regulating the effector functions of T cells, activating co-stimulatory pathways may improve checkpoint inhibition efficacy, and lead to durable antitumor responses. Here, we review recent advances in our understating of co-stimulatory receptors in cancers, providing the necessary groundwork for the rational design of cancer immunotherapy.
Go to : 
References
1. Marin-Acevedo JA, Soyano AE, Dholaria B, Knutson KL, Lou Y. Cancer immunotherapy beyond immune checkpoint inhibitors. J Hematol Oncol. 2018; 11:8.

2. McLane LM, Abdel-Hakeem MS, Wherry EJ. CD8 T cell exhaustion during chronic viral infection and cancer. Annu Rev Immunol. 2019; 37:457–495.
3. Crawford A, Angelosanto JM, Kao C, Doering TA, Odorizzi PM, Barnett BE, Wherry EJ. Molecular and transcriptional basis of CD4+ T cell dysfunction during chronic infection. Immunity. 2014; 40:289–302.
4. Singer M, Wang C, Cong L, Marjanovic ND, Kowalczyk MS, Zhang H, Nyman J, Sakuishi K, Kurtulus S, Gennert D, et al. A distinct gene module for dysfunction uncoupled from activation in tumor-infiltrating T cells. Cell. 2016; 166:1500–1511. e9.
5. Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016; 352:189–196.
6. Chen L, Flies DB. Molecular mechanisms of T cell costimulation and co-inhibition. Nat Rev Immunol. 2013; 13:227–242.
7. Jenkins MK, Ashwell JD, Schwartz RH. Allogeneic non-T spleen cells restore the responsiveness of normal T cell clones stimulated with antigen and chemically modified antigen-presenting cells. J Immunol. 1988; 140:3324–3330.
8. Mueller DL, Jenkins MK, Schwartz RH. An accessory cell-derived costimulatory signal acts independently of protein kinase C activation to allow T cell proliferation and prevent the induction of unresponsiveness. J Immunol. 1989; 142:2617–2628.
9. Jenkins MK, Taylor PS, Norton SD, Urdahl KB. CD28 delivers a costimulatory signal involved in antigen-specific IL-2 production by human T cells. J Immunol. 1991; 147:2461–2466.
10. Linsley PS, Clark EA, Ledbetter JA. T-cell antigen CD28 mediates adhesion with B cells by interacting with activation antigen B7/BB-1. Proc Natl Acad Sci U S A. 1990; 87:5031–5035.
11. Tian R, Wang H, Gish GD, Petsalaki E, Pasculescu A, Shi Y, Mollenauer M, Bagshaw RD, Yosef N, Hunter T, et al. Combinatorial proteomic analysis of intercellular signaling applied to the CD28 T-cell costimulatory receptor. Proc Natl Acad Sci U S A. 2015; 112:E1594–E1603.
12. Fraser JD, Irving BA, Crabtree GR, Weiss A. Regulation of interleukin-2 gene enhancer activity by the T cell accessory molecule CD28. Science. 1991; 251:313–316.
13. June CH, Ledbetter JA, Gillespie MM, Lindsten T, Thompson CB. T-cell proliferation involving the CD28 pathway is associated with cyclosporine-resistant interleukin 2 gene expression. Mol Cell Biol. 1987; 7:4472–4481.
14. Esensten JH, Helou YA, Chopra G, Weiss A, Bluestone JA. CD28 costimulation: from mechanism to therapy. Immunity. 2016; 44:973–988.
15. Strioga M, Pasukoniene V, Characiejus D. CD8+ CD28− and CD8+ CD57+ T cells and their role in health and disease. Immunology. 2011; 134:17–32.
16. Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, Sasmal DK, Huang J, Kim JM, Mellman I, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science. 2017; 355:1428–1433.
17. Rudd CE, Taylor A, Schneider H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol Rev. 2009; 229:12–26.
18. Tsukishiro T, Donnenberg AD, Whiteside TL. Rapid turnover of the CD8+ CD28− T-cell subset of effector cells in the circulation of patients with head and neck cancer. Cancer Immunol Immunother. 2003; 52:599–607.
19. Filaci G, Fenoglio D, Fravega M, Ansaldo G, Borgonovo G, Traverso P, Villaggio B, Ferrera A, Kunkl A, Rizzi M, et al. CD8+ CD28− T regulatory lymphocytes inhibiting T cell proliferative and cytotoxic functions infiltrate human cancers. J Immunol. 2007; 179:4323–4334.
20. Meloni F, Morosini M, Solari N, Passadore I, Nascimbene C, Novo M, Ferrari M, Cosentino M, Marino F, Pozzi E, et al. Foxp3 expressing CD4+ CD25+ and CD8+ CD28− T regulatory cells in the peripheral blood of patients with lung cancer and pleural mesothelioma. Hum Immunol. 2006; 67:1–12.
21. Kamphorst AO, Wieland A, Nasti T, Yang S, Zhang R, Barber DL, Konieczny BT, Daugherty CZ, Koenig L, Yu K, et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science. 2017; 355:1423–1427.
22. Casado JG, Soto R, DelaRosa O, Peralbo E, del Carmen Muñoz-Villanueva M, Rioja L, Peña J, Solana R, Tarazona R. CD8 T cells expressing NK associated receptors are increased in melanoma patients and display an effector phenotype. Cancer Immunol Immunother. 2005; 54:1162–1171.
23. Huff WX, Kwon JH, Henriquez M, Fetcko K, Dey M. The evolving role of CD8+ CD28− immunosenescent T cells in cancer immunology. Int J Mol Sci. 2019; 20:2810.
24. Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, Panoskaltsis N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. 2006; 355:1018–1028.
25. Tyrsin D, Chuvpilo S, Matskevich A, Nemenov D, Römer PS, Tabares P, Hünig T. From TGN1412 to TAB08: the return of CD28 superagonist therapy to clinical development for the treatment of rheumatoid arthritis. Clin Exp Rheumatol. 2016; 34:45–48.
26. Tang XY, Sun Y, Zhang A, Hu GL, Cao W, Wang DH, Zhang B, Chen H. Third-generation CD28/4–1BB chimeric antigen receptor T cells for chemotherapy relapsed or refractory acute lymphoblastic leukaemia: a non-randomised, open-label phase I trial protocol. BMJ Open. 2016; 6:e013904.
27. Cabo M, Offringa R, Zitvogel L, Kroemer G, Muntasell A, Galluzzi L. Trial Watch: immunostimulatory monoclonal antibodies for oncological indications. OncoImmunology. 2017; 6:e1371896.
28. Hutloff A, Dittrich AM, Beier KC, Eljaschewitsch B, Kraft R, Anagnostopoulos I, Kroczek RA. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature. 1999; 397:263–266.
29. McAdam AJ, Chang TT, Lumelsky AE, Greenfield EA, Boussiotis VA, Duke-Cohan JS, Chernova T, Malenkovich N, Jabs C, Kuchroo VK, et al. Mouse inducible costimulatory molecule (ICOS) expression is enhanced by CD28 costimulation and regulates differentiation of CD4+ T cells. J Immunol. 2000; 165:5035–5040.
30. Peach RJ, Bajorath J, Brady W, Leytze G, Greene J, Naemura J, Linsley PS. Complementarity determining region 1 (CDR1)- and CDR3-analogous regions in CTLA-4 and CD28 determine the binding to B7–1. J Exp Med. 1994; 180:2049–2058.
31. Wikenheiser DJ, Stumhofer JS. ICOS costimulation: friend or foe? Front Immunol. 2016; 7:304.
32. McHugh RS, Whitters MJ, Piccirillo CA, Young DA, Shevach EM, Collins M, Byrne MC. CD4+ CD25+ immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity. 2002; 16:311–323.
33. Tafuri A, Shahinian A, Bladt F, Yoshinaga SK, Jordana M, Wakeham A, Boucher LM, Bouchard D, Chan VS, Duncan G, et al. ICOS is essential for effective T-helper-cell responses. Nature. 2001; 409:105–109.
34. Le KS, Amé-Thomas P, Tarte K, Gondois-Rey F, Granjeaud S, Orlanducci F, Foucher ED, Broussais F, Bouabdallah R, Fest T, et al. CXCR5 and ICOS expression identifies a CD8 T-cell subset with T FH features in Hodgkin lymphomas. Blood Adv. 2018; 2:1889–1900.
35. Metzger TC, Long H, Potluri S, Pertel T, Bailey-Bucktrout SL, Lin JC, Fu T, Sharma P, Allison JP, Feldman RM. ICOS promotes the function of CD4+ effector T cells during anti-OX40-mediated tumor rejection. Cancer Res. 2016; 76:3684–3689.
36. Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature. 2016; 537:417–421.
37. Beyrend G, van der Gracht E, Yilmaz A, van Duikeren S, Camps M, Höllt T, Vilanova A, van Unen V, Koning F, de Miranda NF, et al. PD-L1 blockade engages tumor-infiltrating lymphocytes to co-express targetable activating and inhibitory receptors. J Immunother Cancer. 2019; 7:217.
38. Kamphorst AO, Pillai RN, Yang S, Nasti TH, Akondy RS, Wieland A, Sica GL, Yu K, Koenig L, Patel NT, et al. Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc Natl Acad Sci U S A. 2017; 114:4993–4998.
39. Zhang Y, Luo Y, Qin SL, Mu YF, Qi Y, Yu MH, Zhong M. The clinical impact of ICOS signal in colorectal cancer patients. OncoImmunology. 2016; 5:e1141857.
40. Soldevilla MM, Villanueva H, Meraviglia-Crivelli D, Menon AP, Ruiz M, Cebollero J, Villalba M, Moreno B, Lozano T, Llopiz D, et al. ICOS costimulation at the tumor site in combination with ctla-4 blockade therapy elicits strong tumor immunity. Mol Ther. 2019; 27:1878–1891.
41. Fan X, Quezada SA, Sepulveda MA, Sharma P, Allison JP. Engagement of the ICOS pathway markedly enhances efficacy of CTLA-4 blockade in cancer immunotherapy. J Exp Med. 2014; 211:715–725.
42. Chen Q, Mo L, Cai X, Wei L, Xie Z, Li H, Li J, Hu Z. ICOS signal facilitates Foxp3 transcription to favor suppressive function of regulatory T cells. Int J Med Sci. 2018; 15:666–673.
43. Amatore F, Gorvel L, Olive D. Inducible co-stimulator (ICOS) as a potential therapeutic target for anti-cancer therapy. Expert Opin Ther Targets. 2018; 22:343–351.
44. Burns GF, Triglia T, Werkmeister JA, Begley CG, Boyd AW. TLiSA1, a human T lineage-specific activation antigen involved in the differentiation of cytotoxic T lymphocytes and anomalous killer cells from their precursors. J Exp Med. 1985; 161:1063–1078.
45. Gilfillan S, Chan CJ, Cella M, Haynes NM, Rapaport AS, Boles KS, Andrews DM, Smyth MJ, Colonna M. DNAM-1 promotes activation of cytotoxic lymphocytes by nonprofessional antigen-presenting cells and tumors. J Exp Med. 2008; 205:2965–2973.
46. Hou S, Ge K, Zheng X, Wei H, Sun R, Tian Z. CD226 protein is involved in immune synapse formation and triggers natural killer (NK) cell activation via its first extracellular domain. J Biol Chem. 2014; 289:6969–6977.
47. Chan CJ, Andrews DM, Smyth MJ. Receptors that interact with nectin and nectin-like proteins in the immunosurveillance and immunotherapy of cancer. Curr Opin Immunol. 2012; 24:246–251.
48. Lozano E, Dominguez-Villar M, Kuchroo V, Hafler DA. The TIGIT/CD226 axis regulates human T cell function. J Immunol. 2012; 188:3869–3875.
49. Ramsbottom KM, Hawkins ED, Shimoni R, McGrath M, Chan CJ, Russell SM, Smyth MJ, Oliaro J. Cutting edge: DNAX accessory molecule 1-deficient CD8+ T cells display immunological synapse defects that impair antitumor immunity. J Immunol. 2014; 192:553–557.
50. Johnston RJ, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y, Park S, Javinal V, Chiu H, Irving B, et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8+ T cell effector function. Cancer Cell. 2014; 26:923–937.
51. Chauvin JM, Pagliano O, Fourcade J, Sun Z, Wang H, Sander C, Kirkwood JM, Chen TH, Maurer M, Korman AJ, et al. TIGIT and PD-1 impair tumor antigen-specific CD8+ T cells in melanoma patients. J Clin Invest. 2015; 125:2046–2058.
52. Josefsson SE, Huse K, Kolstad A, Beiske K, Pende D, Steen CB, Inderberg EM, Lingjærde OC, Østenstad B, Smeland EB, et al. T cells expressing checkpoint receptor TIGIT are enriched in follicular lymphoma tumors and characterized by reversible suppression of T-cell receptor signaling. Clin Cancer Res. 2018; 24:870–881.
53. Guillerey C, Harjunpää H, Carrié N, Kassem S, Teo T, Miles K, Krumeich S, Weulersse M, Cuisinier M, Stannard K, et al. TIGIT immune checkpoint blockade restores CD8+ T-cell immunity against multiple myeloma. Blood. 2018; 132:1689–1694.
54. Fourcade J, Sun Z, Chauvin JM, Ka M, Davar D, Pagliano O, Wang H, Saada S, Menna C, Amin R, et al. CD226 opposes TIGIT to disrupt Tregs in melanoma. JCI Insight. 2018; 3:e121157.
55. Guillerey C, Ferrari de Andrade L, Vuckovic S, Miles K, Ngiow SF, Yong MC, Teng MW, Colonna M, Ritchie DS, Chesi M, et al. Immunosurveillance and therapy of multiple myeloma are CD226 dependent. J Clin Invest. 2015; 125:2077–2089.
56. Kane LP. T cell Ig and mucin domain proteins and immunity. J Immunol. 2010; 184:2743–2749.
57. Du P, Xiong R, Li X, Jiang J. Immune regulation and antitumor effect of TIM-1. J Immunol Res. 2016; 2016:8605134.
58. Meyers JH, Chakravarti S, Schlesinger D, Illes Z, Waldner H, Umetsu SE, Kenny J, Zheng XX, Umetsu DT, DeKruyff RH, et al. TIM-4 is the ligand for TIM-1, and the TIM-1-TIM-4 interaction regulates T cell proliferation. Nat Immunol. 2005; 6:455–464.
59. Kobayashi N, Karisola P, Peña-Cruz V, Dorfman DM, Jinushi M, Umetsu SE, Butte MJ, Nagumo H, Chernova I, Zhu B, et al. TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity. 2007; 27:927–940.
60. Degauque N, Mariat C, Kenny J, Zhang D, Gao W, Vu MD, Alexopoulos S, Oukka M, Umetsu DT, DeKruyff RH, et al. Immunostimulatory Tim-1-specific antibody deprograms Tregs and prevents transplant tolerance in mice. J Clin Invest. 2008; 118:735–741.
61. Xiao S, Zhu B, Jin H, Zhu C, Umetsu DT, DeKruyff RH, Kuchroo VK. Tim-1 stimulation of dendritic cells regulates the balance between effector and regulatory T cells. Eur J Immunol. 2011; 41:1539–1549.
62. Musgrave BL, Watson CL, Hoskin DW. CD2-CD48 interactions promote cytotoxic T lymphocyte induction and function: anti-CD2 and anti-CD48 antibodies impair cytokine synthesis, proliferation, target recognition/adhesion, and cytotoxicity. J Interferon Cytokine Res. 2003; 23:67–81.
63. Wild MK, Strittmatter W, Matzku S, Schraven B, Meuer SC. Tumor therapy with bispecific antibody: the targeting and triggering steps can be separated employing a CD2-based strategy. J Immunol. 1999; 163:2064–2072.
64. Cheadle EJ, Rothwell DG, Bridgeman JS, Sheard VE, Hawkins RE, Gilham DE. Ligation of the CD2 co-stimulatory receptor enhances IL-2 production from first-generation chimeric antigen receptor T cells. Gene Ther. 2012; 19:1114–1120.
65. Valdez PA, Wang H, Seshasayee D, van Lookeren Campagne M, Gurney A, Lee WP, Grewal IS. NTB-A, a new activating receptor in T cells that regulates autoimmune disease. J Biol Chem. 2004; 279:18662–18669.
66. Flaig RM, Stark S, Watzl C. Cutting edge: NTB-A activates NK cells via homophilic interaction. J Immunol. 2004; 172:6524–6527.
67. Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, Yates KB, Lako A, Felt K, Naik GS, et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol. 2019; 20:326–336.
68. Eisenberg G, Engelstein R, Geiger-Maor A, Hajaj E, Merims S, Frankenburg S, Uzana R, Rutenberg A, Machlenkin A, Frei G, et al. Soluble SLAMF6 receptor induces strong CD8+ T-cell effector function and improves anti-melanoma activity in vivo. Cancer Immunol Res. 2018; 6:127–138.
69. Goodwin RG, Din WS, Davis-Smith T, Anderson DM, Gimpel SD, Sato TA, Maliszewski CR, Brannan CI, Copeland NG, Jenkins NA, et al. Molecular cloning of a ligand for the inducible T cell gene 4–1BB: a member of an emerging family of cytokines with homology to tumor necrosis factor. Eur J Immunol. 1993; 23:2631–2641.
70. Vinay DS, Kwon BS. Role of 4–1BB in immune responses. Semin Immunol. 1998; 10:481–489.
71. Menk AV, Scharping NE, Rivadeneira DB, Calderon MJ, Watson MJ, Dunstane D, Watkins SC, Delgoffe GM. 4–1BB costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses. J Exp Med. 2018; 215:1091–1100.
72. Chester C, Sanmamed MF, Wang J, Melero I. Immunotherapy targeting 4–1BB: mechanistic rationale, clinical results, and future strategies. Blood. 2018; 131:49–57.
73. Chester C, Ambulkar S, Kohrt HE. 4–1BB agonism: adding the accelerator to cancer immunotherapy. Cancer Immunol Immunother. 2016; 65:1243–1248.
74. Drenkard D, Becke FM, Langstein J, Spruss T, Kunz-Schughart LA, Tan TE, Lim YC, Schwarz H. CD137 is expressed on blood vessel walls at sites of inflammation and enhances monocyte migratory activity. FASEB J. 2007; 21:456–463.
75. Teijeira Á, Palazón A, Garasa S, Marré D, Aubá C, Rogel A, Murillo O, Martínez-Forero I, Lang F, Melero I, et al. CD137 on inflamed lymphatic endothelial cells enhances CCL21-guided migration of dendritic cells. FASEB J. 2012; 26:3380–3392.
76. Palazón A, Teijeira A, Martínez-Forero I, Hervás-Stubbs S, Roncal C, Peñuelas I, Dubrot J, Morales-Kastresana A, Pérez-Gracia JL, Ochoa MC, et al. Agonist anti-CD137 mAb act on tumor endothelial cells to enhance recruitment of activated T lymphocytes. Cancer Res. 2011; 71:801–811.
77. Saoulli K, Lee SY, Cannons JL, Yeh WC, Santana A, Goldstein MD, Bangia N, DeBenedette MA, Mak TW, Choi Y, et al. CD28-independent, TRAF2-dependent costimulation of resting T cells by 4–1BB ligand. J Exp Med. 1998; 187:1849–1862.
78. Wolfl M, Kuball J, Ho WY, Nguyen H, Manley TJ, Bleakley M, Greenberg PD. Activation-induced expression of CD137 permits detection, isolation, and expansion of the full repertoire of CD8+ T cells responding to antigen without requiring knowledge of epitope specificities. Blood. 2007; 110:201–210.
79. Kim HD, Park S, Jeong S, Lee YJ, Lee H, Kim CG, Kim KH, Hong SM, Lee JY, Kim S, et al. 4–1BB delineates distinct activation status of exhausted tumor-infiltrating CD8+ T cells in hepatocellular carcinoma. Hepatology. 2019. DOI: doi: 10.1002/hep.30881.
80. Ye Q, Song DG, Poussin M, Yamamoto T, Best A, Li C, Coukos G, Powell DJ Jr. CD137 accurately identifies and enriches for naturally occurring tumor-reactive T cells in tumor. Clin Cancer Res. 2014; 20:44–55.
81. Thommen DS, Koelzer VH, Herzig P, Roller A, Trefny M, Dimeloe S, Kiialainen A, Hanhart J, Schill C, Hess C, et al. A transcriptionally and functionally distinct PD-1+ CD8+ T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat Med. 2018; 24:994–1004.
82. Williams JB, Horton BL, Zheng Y, Duan Y, Powell JD, Gajewski TF. The EGR2 targets LAG-3 and 4–1BB describe and regulate dysfunctional antigen-specific CD8+ T cells in the tumor microenvironment. J Exp Med. 2017; 214:381–400.
83. Weigelin B, Bolaños E, Teijeira A, Martinez-Forero I, Labiano S, Azpilikueta A, Morales-Kastresana A, Quetglas JI, Wagena E, Sánchez-Paulete AR, et al. Focusing and sustaining the antitumor CTL effector killer response by agonist anti-CD137 mAb. Proc Natl Acad Sci U S A. 2015; 112:7551–7556.
84. Houot R, Goldstein MJ, Kohrt HE, Myklebust JH, Alizadeh AA, Lin JT, Irish JM, Torchia JA, Kolstad A, Chen L, et al. Therapeutic effect of CD137 immunomodulation in lymphoma and its enhancement by Treg depletion. Blood. 2009; 114:3431–3438.
85. Wilcox RA, Flies DB, Zhu G, Johnson AJ, Tamada K, Chapoval AI, Strome SE, Pease LR, Chen L. Provision of antigen and CD137 signaling breaks immunological ignorance, promoting regression of poorly immunogenic tumors. J Clin Invest. 2002; 109:651–659.
86. Mayes PA, Hance KW, Hoos A. The promise and challenges of immune agonist antibody development in cancer. Nat Rev Drug Discov. 2018; 17:509–527.
87. Qi X, Li F, Wu Y, Cheng C, Han P, Wang J, Yang X. Optimization of 4–1BB antibody for cancer immunotherapy by balancing agonistic strength with Fcγ R affinity. Nat Commun. 2019; 10:2141.
88. Hinner MJ, Aiba RS, Jaquin TJ, Berger S, Dürr MC, Schlosser C, Allersdorfer A, Wiedenmann A, Matschiner G, Schüler J, et al. Tumor-localized costimulatory t-cell engagement by the 4–1bb/her2 bispecific antibody-anticalin fusion prs-343. Clin Cancer Res. 2019; 25:5878–5889.
89. Claus C, Ferrara C, Xu W, Sam J, Lang S, Uhlenbrock F, Albrecht R, Herter S, Schlenker R, Hüsser T, et al. Tumor-targeted 4–1BB agonists for combination with T cell bispecific antibodies as off-the-shelf therapy. Sci Transl Med. 2019; 11:eaav5989.
90. Labrijn AF, Janmaat ML, Reichert JM, Parren PW. Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discov. 2019; 18:585–608.
91. Linch SN, McNamara MJ, Redmond WL. Ox40 agonists and combination immunotherapy: Putting the pedal to the metal. Front Oncol. 2015; 5:34.
92. Ishii N, Takahashi T, Soroosh P, Sugamura K. OX40-OX40 ligand interaction in T-cell-mediated immunity and immunopathology. Adv Immunol. 2010; 105:63–98.
93. Sugamura K, Ishii N, Weinberg AD. Therapeutic targeting of the effector T-cell co-stimulatory molecule OX40. Nat Rev Immunol. 2004; 4:420–431.
94. Song J, Salek-Ardakani S, Rogers PR, Cheng M, Van Parijs L, Croft M. The costimulation-regulated duration of PKB activation controls T cell longevity. Nat Immunol. 2004; 5:150–158.
95. Song J, So T, Cheng M, Tang X, Croft M. Sustained survivin expression from OX40 costimulatory signals drives T cell clonal expansion. Immunity. 2005; 22:621–631.
96. Song J, So T, Croft M. Activation of NF-kappaB1 by OX40 contributes to antigen-driven T cell expansion and survival. J Immunol. 2008; 180:7240–7248.
97. Ito T, Wang YH, Duramad O, Hanabuchi S, Perng OA, Gilliet M, Qin FX, Liu YJ. OX40 ligand shuts down IL-10-producing regulatory T cells. Proc Natl Acad Sci U S A. 2006; 103:13138–13143.
98. Willoughby J, Griffiths J, Tews I, Cragg MS. OX40: structure and function – what questions remain? Mol Immunol. 2017; 83:13–22.
99. Kjaergaard J, Tanaka J, Kim JA, Rothchild K, Weinberg A, Shu S. Therapeutic efficacy of OX-40 receptor antibody depends on tumor immunogenicity and anatomic site of tumor growth. Cancer Res. 2000; 60:5514–5521.
100. Linch SN, Kasiewicz MJ, McNamara MJ, Hilgart-Martiszus IF, Farhad M, Redmond WL. Combination OX40 agonism/CTLA-4 blockade with HER2 vaccination reverses T-cell anergy and promotes survival in tumor-bearing mice. Proc Natl Acad Sci U S A. 2016; 113:E319–E327.
101. Ladányi A, Somlai B, Gilde K, Fejös Z, Gaudi I, Tímár J. T-cell activation marker expression on tumor-infiltrating lymphocytes as prognostic factor in cutaneous malignant melanoma. Clin Cancer Res. 2004; 10:521–530.
102. Petty JK, He K, Corless CL, Vetto JT, Weinberg AD. Survival in human colorectal cancer correlates with expression of the T-cell costimulatory molecule OX-40 (CD134). Am J Surg. 2002; 183:512–518.
103. Boettler T, Moeckel F, Cheng Y, Heeg M, Salek-Ardakani S, Crotty S, Croft M, von Herrath MG. OX40 facilitates control of a persistent virus infection. PLoS Pathog. 2012; 8:e1002913.
104. Peng W, Williams LJ, Xu C, Melendez B, McKenzie JA, Chen Y, Jackson HL, Voo KS, Mbofung RM, Leahey SE, et al. Anti-OX40 antibody directly enhances the function of tumor-reactive CD8+ T cells and synergizes with PI3Kβ inhibition in PTEN loss melanoma. Clin Cancer Res. 2019; 25:6406–6416.
105. Curti BD, Kovacsovics-Bankowski M, Morris N, Walker E, Chisholm L, Floyd K, Walker J, Gonzalez I, Meeuwsen T, Fox BA, et al. OX40 is a potent immune-stimulating target in late-stage cancer patients. Cancer Res. 2013; 73:7189–7198.
106. Piconese S, Valzasina B, Colombo MP. OX40 triggering blocks suppression by regulatory T cells and facilitates tumor rejection. J Exp Med. 2008; 205:825–839.
107. Polesso F, Sarker M, Weinberg AD, Murray SE, Moran AE. OX40 agonist tumor immunotherapy does not impact regulatory T cell suppressive function. J Immunol. 2019; 203:2011–2019.
108. Redmond WL, Linch SN, Kasiewicz MJ. Combined targeting of costimulatory (OX40) and coinhibitory (CTLA-4) pathways elicits potent effector T cells capable of driving robust antitumor immunity. Cancer Immunol Res. 2014; 2:142–153.
109. Guo Z, Wang X, Cheng D, Xia Z, Luan M, Zhang S. PD-1 blockade and OX40 triggering synergistically protects against tumor growth in a murine model of ovarian cancer. PLoS One. 2014; 9:e89350.
110. Shrimali RK, Ahmad S, Verma V, Zeng P, Ananth S, Gaur P, Gittelman RM, Yusko E, Sanders C, Robins H, et al. Concurrent pd-1 blockade negates the effects of OX40 agonist antibody in combination immunotherapy through inducing T-cell apoptosis. Cancer Immunol Res. 2017; 5:755–766.
111. Redmond WL, Triplett T, Floyd K, Weinberg AD. Dual anti-OX40/IL-2 therapy augments tumor immunotherapy via IL-2R-mediated regulation of OX40 expression. PLoS One. 2012; 7:e34467.
112. Kvarnhammar AM, Veitonmäki N, Hägerbrand K, Dahlman A, Smith KE, Fritzell S, von Schantz L, Thagesson M, Werchau D, Smedenfors K, et al. The CTLA-4 x OX40 bispecific antibody ATOR-1015 induces anti-tumor effects through tumor-directed immune activation. J Immunother Cancer. 2019; 7:103.
113. Knee DA, Hewes B, Brogdon JL. Rationale for anti-GITR cancer immunotherapy. Eur J Cancer. 2016; 67:1–10.
114. Nocentini G, Riccardi C. GITR: a modulator of immune response and inflammation. Adv Exp Med Biol. 2009; 647:156–173.
115. Snell LM, McPherson AJ, Lin GH, Sakaguchi S, Pandolfi PP, Riccardi C, Watts TH. CD8 T cell-intrinsic GITR is required for T cell clonal expansion and mouse survival following severe influenza infection. J Immunol. 2010; 185:7223–7234.
116. Kim IK, Kim BS, Koh CH, Seok JW, Park JS, Shin KS, Bae EA, Lee GE, Jeon H, Cho J, et al. Glucocorticoid-induced tumor necrosis factor receptor-related protein costimulation facilitates tumor regression by inducing IL-9-producing helper T cells. Nat Med. 2015; 21:1010–1017.
117. Zhan Y, Gerondakis S, Coghill E, Bourges D, Xu Y, Brady JL, Lew AM. Glucocorticoid-induced TNF receptor expression by T cells is reciprocally regulated by NF-kappaB and NFAT. J Immunol. 2008; 181:5405–5413.
118. Clouthier DL, Zhou AC, Wortzman ME, Luft O, Levy GA, Watts TH. GITR intrinsically sustains early type 1 and late follicular helper CD4 T cell accumulation to control a chronic viral infection. PLoS Pathog. 2015; 11:e1004517.
119. Vence L, Bucktrout SL, Fernandez Curbelo I, Blando J, Smith BM, Mahne AE, Lin JC, Park T, Pascua E, Sai T, et al. Characterization and comparison of GITR expression in solid tumors. Clin Cancer Res. 2019; 25:6501–6510.
120. Sabharwal SS, Rosen DB, Grein J, Tedesco D, Joyce-Shaikh B, Ueda R, Semana M, Bauer M, Bang K, Stevenson C, et al. GITR agonism enhances cellular metabolism to support CD8+ T-cell proliferation and effector cytokine production in a mouse tumor model. Cancer Immunol Res. 2018; 6:1199–1211.
121. Cho JS, Hsu JV, Morrison SL. Localized expression of GITR-L in the tumor microenvironment promotes CD8+ T cell dependent anti-tumor immunity. Cancer Immunol Immunother. 2009; 58:1057–1069.
122. Cohen AD, Schaer DA, Liu C, Li Y, Hirschhorn-Cymmerman D, Kim SC, Diab A, Rizzuto G, Duan F, Perales MA, et al. Agonist anti-GITR monoclonal antibody induces melanoma tumor immunity in mice by altering regulatory T cell stability and intra-tumor accumulation. PLoS One. 2010; 5:e10436.
123. Kim YH, Shin SM, Choi BK, Oh HS, Kim CH, Lee SJ, Kim KH, Lee DG, Park SH, Kwon BS. Authentic GITR signaling fails to induce tumor regression unless Foxp3+ regulatory T cells are depleted. J Immunol. 2015; 195:4721–4729.
124. Wang B, Zhang W, Jankovic V, Golubov J, Poon P, Oswald EM, Gurer C, Wei J, Ramos I, Wu Q, et al. Combination cancer immunotherapy targeting PD-1 and GITR can rescue CD8+ T cell dysfunction and maintain memory phenotype. Sci Immunol. 2018; 3:eaat7061.
125. Narumi K, Miyakawa R, Shibasaki C, Henmi M, Mizoguchi Y, Ueda R, Hashimoto H, Hiraoka N, Yoshida T, Aoki K. Local administration of GITR agonistic antibody induces a stronger antitumor immunity than systemic delivery. Sci Rep. 2019; 9:5562.
126. Ahrends T, Spanjaard A, Pilzecker B, Babala N, Bovens A, Xiao Y, Jacobs H, Borst J. CD4+ T cell help confers a cytotoxic T cell effector program including coinhibitory receptor downregulation and increased tissue invasiveness. Immunity. 2017; 47:848–861. e5.
127. Keller AM, Schildknecht A, Xiao Y, van den Broek M, Borst J. Expression of costimulatory ligand CD70 on steady-state dendritic cells breaks CD8+ T cell tolerance and permits effective immunity. Immunity. 2008; 29:934–946.
128. Claus C, Riether C, Schürch C, Matter MS, Hilmenyuk T, Ochsenbein AF. CD27 signaling increases the frequency of regulatory T cells and promotes tumor growth. Cancer Res. 2012; 72:3664–3676.
129. Wasiuk A, Testa J, Weidlick J, Sisson C, Vitale L, Widger J, Crocker A, Thomas LJ, Goldstein J, Marsh HC, et al. CD27-mediated regulatory T cell depletion and effector T cell costimulation both contribute to antitumor efficacy. J Immunol. 2017; 199:4110–4123.
130. He LZ, Prostak N, Thomas LJ, Vitale L, Weidlick J, Crocker A, Pilsmaker CD, Round SM, Tutt A, Glennie MJ, et al. Agonist anti-human CD27 monoclonal antibody induces T cell activation and tumor immunity in human CD27-transgenic mice. J Immunol. 2013; 191:4174–4183.
131. Ward-Kavanagh LK, Lin WW, Šedý JR, Ware CF. The TNF receptor superfamily in co-stimulating and co-inhibitory responses. Immunity. 2016; 44:1005–1019.
132. Twohig JP, Marsden M, Cuff SM, Ferdinand JR, Gallimore AM, Perks WV, Al-Shamkhani A, Humphreys IR, Wang EC. The death receptor 3/TL1A pathway is essential for efficient development of antiviral CD4+ and CD8+ T-cell immunity. FASEB J. 2012; 26:3575–3586.
133. Meylan F, Richard AC, Siegel RM. TL1A and DR3, a TNF family ligand-receptor pair that promotes lymphocyte costimulation, mucosal hyperplasia, and autoimmune inflammation. Immunol Rev. 2011; 244:188–196.
134. Richard AC, Tan C, Hawley ET, Gomez-Rodriguez J, Goswami R, Yang XP, Cruz AC, Penumetcha P, Hayes ET, Pelletier M, et al. The TNF-family ligand TL1A and its receptor DR3 promote T cell-mediated allergic immunopathology by enhancing differentiation and pathogenicity of IL-9-producing T cells. J Immunol. 2015; 194:3567–3582.
135. Tam EM, Fulton RB, Sampson JF, Muda M, Camblin A, Richards J, Koshkaryev A, Tang J, Kurella V, Jiao Y, et al. Antibody-mediated targeting of TNFR2 activates CD8+ T cells in mice and promotes antitumor immunity. Sci Transl Med. 2019; 11:eaax0720.
Go to :
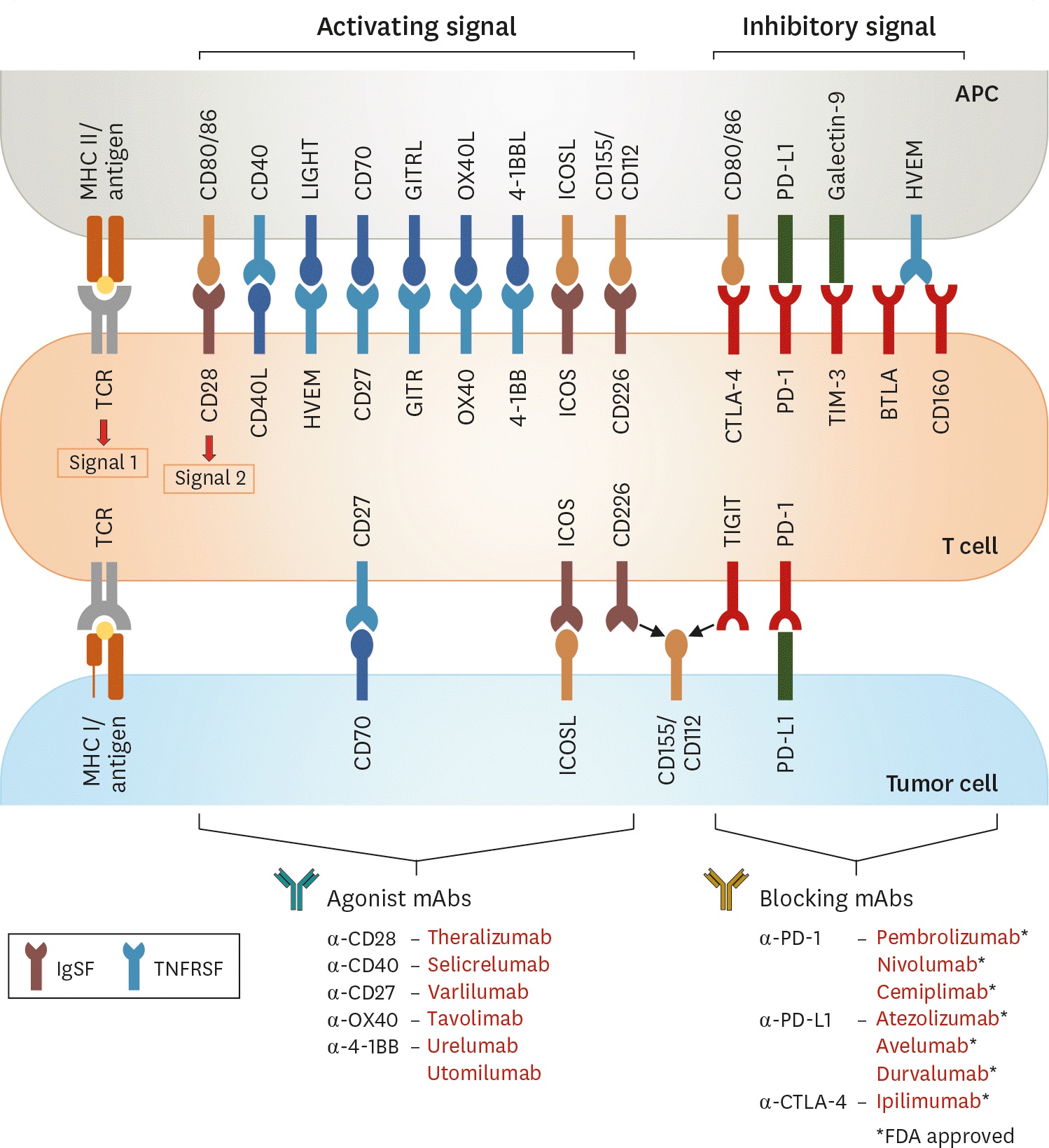 | Figure 1.Schematic overview of co-stimulatory/inhibitory receptors expressed by T cells interacting with their counterpart on APCs or tumor cells. Inhibitory and stimulatory receptors expressed on T cells in the tumor microenvironment may be targeted for therapeutic intervention by development of agonist targeting co-stimulatory receptors and/or blocking Abs targeting immune inhibitory receptors. CD40L, CD40 ligand; LIGHT, lymphocyte activation gene 3 protein; HVEM, herpes virus-entry mediator; BTLA, B-lymphocyte and T-lymphocyte attenuator. |
 XML Download
XML Download