

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Despite a risen awareness among the clinical society, diabetes mellitus (DM) remains a growing epidemic which affected 425 million adults in 2017, and this number is expected to increase to 629 million individuals affected by DM until 2045 (www.idf.org). DM has a huge impact on the prognosis of these patients through the traditional accompanying macro- and microvascular complications which are leading to a markedly increased morbidity and mortality. Since the Framingham Heart Study, it is known that DM also increases the incidence of heart failure (HF), which is mainly related to accelerated atherosclerosis and an increased incidence and severity of myocardial infarction, as well as the frequent coexistence of arterial hypertension in diabetic individuals. However, the risk for HF is also increased in an underestimated amount of diabetic patients in the absence of these risk factors, an entity termed diabetic cardiomyopathy (DbCM) [1]. Given that many molecular alterations and mechanisms identified in failing hearts are also similarly altered in DbCM, DbCM can be considered a predisposition to develop cardiac dysfunction, in particular in the presence of other stress factors such as ischemic heart disease or hypertension. Mitochondria are the powerhouse of the cell, continuously providing large amounts of adenosine triphosphate (ATP) to cardiomyocytes which require an extraordinary ATP turnover to maintain contractile function. Of note, not only in HF but also in DbCM, a number of defects in mitochondrial biology have been consistently reported, and a variety of distinct mechanisms have been proposed to lead to these mitochondrial defects [2]. Nevertheless, the underlying mechanisms of mitochondrial defects remain incompletely understood, and with increasing understanding of mitochondrial biology in recent years, a number of novel pathways and mechanisms have been elucidated that may contribute to mitochondrial dysfunction in the diabetic heart. In the current review, we will discuss established and novel mechanisms of myocardial mitochondrial dysfunction in DM.
DEFINITION AND CLINICAL PHENOTYPE OF DbCM
The first evidence supporting the existence of a DbCM has been published by Rubler et al. [1] in 1972 who identified four patients in a post-mortem analysis that died from HF, had suffered from DM, but had no other known etiology of HF. Subsequently, the Framingham Heart Study reported a significantly increased incidence of HF in patients with DM, with a 2-fold increase in men and a 5-fold increase in women [3]. Besides many more studies confirming the increased risk for HF in diabetics, DM has also been shown to be over-represented in patients hospitalized for HF, and the prognosis of HF in type 2 diabetes mellitus (T2DM) patients is worse than in patients with HF alone [4567]. These observations have led to the hypothesis that diabetic patients may develop molecular alterations affecting the heart independently of the macrovascular complications like coronary artery disease (CAD) or hypertension which are also driven by DM [8]. Today, the definition of DbCM describes a cardiac dysfunction in diabetic individuals in the absence of CAD, hypertension, valvular heart disease, congenital heart disease, or any other known etiology of cardiomyopathy [9].
The clinical phenotype of DbCM has been a subject of debate. The typical traits of DbCM are thought to include cardiac hypertrophy and diastolic dysfunction, which may become clinically manifest as heart failure with preserved ejection fraction (HFpEF) [910]. In addition, a number of studies reported subclinical systolic dysfunction in patients with DbCM detected by echocardiographic strain analysis. Some authors think of these two phenotypes as one continuum where DbCM progresses from the HFpEF phenotype with cardiac hypertrophy and concomitant diastolic dysfunction to a later stage with further structural damage, leading to a rather dilated phenotype with systolic impairment [11]. Others suppose that the two phenotypes may be different entities with distinct mechanisms contributing to either one [1012]. Longitudinal data observing the evolution of DbCM in a well-defined patient cohort with exclusion of confounding pathologies would be needed to shed more light on this issue.
MITOCHONDRIAL DYSFUNCTION IN DbCM
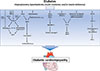
Numerous mechanisms have been identified and proposed to contribute to the pathogenesis of DbCM, including oxidative stress, fibrosis, inflammation, various forms of cell death, alterations in myocardial energetics, among others [2]. Of particular importance may be the development of mitochondrial dysfunction, which may not only contribute to the development of diabetic complications in different organs, but may also contribute to the development of T2DM by inducing insulin resistance in skeletal muscle, adipose tissue and pancreatic β-cells [13]. In rodents models of DM, which are per se resistant to the development of CAD and hypertension, defects in mitochondrial biology have been observed already several decades ago. As early as 1985, an impairment in state 3 respiration of isolated mitochondria has been reported in hearts of obese or diabetic mice [14]. Ever since, mitochondrial dysfunction has been observed in numerous rodent models of DM, including models of type 1 diabetes mellitus (T1DM) such as streptozotocin (STZ)-diabetic rodents, OVE26 mice, non-obese diabetic (NOD) mice, or in the Akita mouse model, as well as models of T2DM such as ob/ob mice, db/db mice, Zucker (diabetic) fatty rats, Goto Kakizaki rats, and in models of diet-induced obesity [15]. In humans, mitochondrial dysfunction was observed in atrial tissue of DM patients by Anderson et al. [16] who demonstrated impaired respiration rates of isolated mitochondria using fatty acids (FAs) or glutamate as a substrate, and increased generation of hydrogen peroxide (H2O2). Furthermore, studies using atrial tissue or tissue of atrial appendage also reported impaired respiration rates and electron transport chain (ETC) complex activities in diabetic individuals [1718]. Taken together, there is compelling evidence that alterations in mitochondrial function exist in rodent and human DbCM. Underlying mechanisms of impaired mitochondrial biology in DbCM will be discussed in the following sections (Fig. 1).
MITOCHONDRIAL MECHANISMS OF DbCM
Altered mitochondrial substrate utilization
To maintain continuous pump function, the heart requires large amounts of high energy phosphates and accounts for approximately 8% of the total ATP consumption of the body. The vast majority of this ATP is regenerated in the mitochondria via oxidative phosphorylation (OXPHOS), which explains the high mitochondrial volume density of 30% to 40% in the heart, dependent on the species [19]. In the absence of DM or other cardiac pathologies, the majority of ATP is derived from the oxidation of FAs (60% to 70%), whereas a minor part is derived from the oxidation of glucose, lactate, ketone bodies, and amino acids (20% to 30%), depending on their availability in the blood [20212223]. The resulting reducing equivalents (NADH, FADH2) deliver electrons into the ETC, where electrons are transported through the distinct complexes of the ETC and finally transferred onto molecular oxygen by the activity of complex IV, thereby reducing O2 to H2O. This electron transport is used by the ETC complexes to build up an electrochemical gradient by pumping protons into the intermembranous space. The energy released by back flow of protons into the mitochondrial matrix via the FO subunit of the FOF1-ATPase is used by the FOF1-ATPase to regenerate ATP from adenosine diphosphate (ADP); thus, ATP regeneration is coupled to oxygen consumption.
In DM, the typically observed increase in serum FAs and triglycerides promotes an increase in FA uptake and oxidation. Evaluation of myocardial substrate oxidation in isolated working hearts demonstrated increased rates of fatty acid oxidation (FAO) and decreased oxidation of glucose in various animal models of T2DM, including db/db mice, ob/ob mice, or Zucker diabetic fatty rats [2425]. Similar observations have been made in humans, where rates of FA uptake and oxidation were increased and insulin-stimulated glucose uptake and glucose utilization were decreased in insulin-resistant and/or diabetic individuals [26272829]. Increased FAO rates are driven, at least in part, by increased activity of peroxisome proliferator-activated receptors (PPARs), in particular PPARα. Both activation of PPARα by FAs and peroxisome proliferator-activated receptor gamma coactivator 1α (PGC-1α) as well as increased PPARα expression increase the binding of PPARα to PPAR response elements on promoters of genes encoding for proteins involved in FA uptake and oxidation, including carnitine palmitoyltransferase 1 (CPT1) and long chain acyl-CoA dehydrogenase (LCAD) as key enzymes of FAO [30]. Indeed, cardiomyocyte-specific overexpression of PPARα resulted in increased expression of FA utilization genes and increased FAO [31]. In turn, expression of genes encoding for enzymes involved in glucose oxidation was reduced, accompanied by decreased glucose uptake and oxidation. Of note though, PPARα signaling was not increased as early as 4 weeks of age in these mice, whereas FAO rates were increased. Thus, PPARα-independent mechanisms may increase FAO rates in the early disease state, whereas PPARα signaling may sustain elevated rates of FAO in the heart as DM persists.
Increased rates of FAO affect the mitochondrial efficiency of ATP regeneration. Mjos [32] demonstrated more than four decades ago that increasing cardiac FA uptake by lipid infusion leads to enhanced oxygen extraction in healthy dogs. Since cardiac contractility remained unaltered, the ratio of cardiac work to O2 consumption (i.e., cardiac efficiency) was impaired. Similarly, FAO and myocardial O2 consumption are increased and cardiac efficiency is decreased in hearts of ob/ob and db/db mice, as well as in humans with obesity and insulin resistance [242733]. The mechanism of increased O2 consumption may be caused by mitochondrial uncoupling. Boudina et al. [3334] demonstrated that the presence of long chain FA in the perfusion medium during Langendorff perfusion of db/db hearts increased O2 consumption and impaired the ATP/O ratio, indicating FA-induced mitochondrial uncoupling. The proton leak could be inhibited by guanosine triphosphate (GTP), which blocks uncoupling protein (UCP) activity. Given an increase in mitochondrial ROS, it has been proposed that increased mitochondrial ROS would directly activate mitochondrial UCPs, thereby increasing oxygen consumption and impairing cardiac contractility due to decreased ATP regeneration, resulting in impaired cardiac efficiency. A small component of the proton leak in db/db heart mitochondria was also sensitive to inhibition with atractyloside, suggesting some uncoupling to be mediated also by activity of the adenine nucleotide translocase. Of note, mice lacking insulin receptors in cardiomyocytes (CIRKO mice) also showed impaired ATP/O ratios when respiring palmitoyl-carnitine, which was normalized by scavenging mitochondrial ROS using manganese (III) tetrakis (4-benzoic acid) porphyrin (MnTBAP), thereby not only confirming that ROS may induce uncoupling but also suggesting a critical role of cardiac insulin resistance in the pathogenesis of ROS-induced mitochondrial uncoupling [35]. Furthermore, increasing FA delivery to CIRKO hearts by induction of STZ-dependent DM was a prerequisite to develop increased O2 consumption and impaired cardiac efficiency [36]. Given that CIRKO mice displayed marked proteomic remodeling with downregulation of FAO proteins, it was proposed that overwhelming the FAO capacity of CIRKO mice by additional hyperlipidemia may have contributed to induction of uncoupling and cardiac inefficiency. Importantly, hearts of ob/ob and db/db mice display cardiomyocyte insulin resistance when evaluated by insulin-stimulated glucose uptake and glycolysis [37]. Thus, it can be speculated that insulin-resistant cardiomyocytes in T2DM hearts are susceptible to FA-induced ROS-mediated activation of UCPs, thereby impairing cardiac efficiency.
If the delivery of FA to mitochondria exceeds the mitochondrial capacity for FAO, acyl-CoAs accumulate and are directed into other pathways of lipid metabolism such as synthesis of triacylglycerols (TAG), ceramides, diacylglycerols, and acyl-carnitines [38]. Not only in many animal models of diabetes, but also in human diabetic subjects, diabetes drives accumulation of TAG in the heart [3940]. Recent lipidomics profiling also demonstrated a pronounced remodeling of the myocardial phospholipid pool in rat DbCM [41]. While storage of FA as TAG may serve a protective function to prevent excessive generation of reactive lipid intermediates, ceramide and diacylglycerol accumulation might contribute to the development of lipotoxic cardiac dysfunction, e.g., by activation of protein kinase C signaling, apoptosis, endoplasmic reticulum (ER) stress, and increased ROS generation [124243]. Levels of ceramides can be markedly increased in obese and diabetic rats, and inhibition of the rate-limiting enzyme of ceramide biosynthesis, serine palmitoyltransferase, resulted in improved systolic function in isolated hearts of mice with lipotoxic cardiomyopathy [44]. Accumulation of acyl-CoAs may also be detrimental to the diabetic heart, given that increased cardiomyocyte levels of acyl-CoA in mice overexpressing long-chain acyl-CoA synthetase 1 (ACSL1) show increased apoptosis and ceramide levels, marked fragmentation of mitochondria, increased mitochondrial ROS generation, and defects in cardiac contractility [4546]. While the role of phospholipid remodeling in DbCM remains unresolved, it is tempting to speculate that mitochondrial membrane remodeling could potentially alter OXPHOS function, ROS generation and the process of mitochondrial fusion and fission.
Another effect of FA on the myocardium in diabetes relates to the cardiac adaptation to hypoxic conditions [47]. Work from the Heather lab showed that diabetic hearts fail to accumulate hypoxia-inducible factor (HIF)-1α during ischemia. Using a model of insulin resistance in HL-1 cardiomyocytes, they showed that long-chain FA may prevent HIF-1α accumulation and subsequent protective HIF-1α downstream signaling due to prevention of succinate accumulation, suggesting that increased FA levels in diabetes may impair cardiac HIF-1α accumulation and subsequent cardioprotection [47].
A cytokine-related mechanism that may cause mitochondrial dysfunction and uncoupling in diabetic hearts is impaired cardiac adiponectin action. Adiponectin is an adipose-derived cytokine, whose levels are decreased in obese and diabetic individuals, and hypoadiponectinemia has been established as an independent risk factor for cardiovascular disease [48]. Adiponectin deficiency has been shown to impair mitochondrial function in various tissues, and normalizing serum adiponectin levels in ob/ob mice attenuated defects in ETC complex activities [49505152]. These effects of adiponectin are likely mediated via adiponectin receptor 1 (AdipoR1), the main receptor for adiponectin expressed in cardiomyocytes. Similar to adiponectin deficiency, deletion of AdipoR1 in skeletal muscle compromised mitochondrial biogenesis and ETC complex activities by suppressing the signaling axis of AMP-activated protein kinase (AMPK)-sirtuin 1 (SIRT1)-PGC-1α [5354]. In the heart, we demonstrated that deletion of AdipoR1 but not AdipoR2 results in impaired ETC complex activities, accompanied by decreased expression of ETC subunits and impaired AMPK-SIRT1-PGC-1α signaling [55]. We also observed decreased ATP/O ratios and an impairment in cardiac efficiency, and both were normalized by mitochondria-targeted ROS scavenging. In addition, inhibition of UCPs with guanosine diphosphate also normalized mitochondrial uncoupling, indicative of ROS-induced uncoupling. Of note though, FAO rates were not increased, suggesting that ROS-induced UCP-mediated uncoupling and impairment in cardiac efficiency may not depend on FAs in this model. Nevertheless, since both serum adiponectin levels and cardiac AdipoR1 expression are reduced in models of diet-induced obesity and DM, impaired AdipoR1 signaling may also contribute to impaired mitochondrial coupling and cardiac efficiency in DbCM (unpublished results) [51].
It remains to be mentioned though that not all animal models of DM (e.g., Zucker diabetic fatty [ZDF] rats, T1DM Akita mice) develop increased O2 consumption, mitochondrial uncoupling or impaired cardiac efficiency [56]. In addition, other mechanisms may contribute to impaired cardiac efficiency, such as FAs being a less efficient fuel for ATP regeneration. Theoretical calculations predict that shifting substrate oxidation from 100% palmitate to 100% glucose would increase ATP yield by 12% to 14%, thus suggesting a higher oxygen cost to produce ATP and thereby also explaining increased O2 consumption and impaired cardiac efficiency. However, since the relative substrate shift in diabetic hearts from glucose to FAs is much less pronounced than in this theoretical calculation, such a mechanisms may only become relevant in a setting of increased energy demand, such as marked hypertension or ischemic insults.
Oxidative stress
One of the established mechanisms contributing to DbCM is oxidative stress through imbalanced generation and scavenging of ROS. Superoxide (O2·−) can be generated by the reduction of molecular oxygen, is highly reactive and a precursor of other species of ROS. H2O2, being a non-radical form of ROS, can result from dismutation of O2·−, either spontaneously or by the activity of superoxide dismutases (SODs). O2·− and H2O2 can generate the hydroxyl radical (·OH) which may have the greatest oxidative potential of all forms of ROS. In the heart, mitochondria may represent the major source of ROS production. Within mitochondria, the main source of ROS is the ETC, both resulting from unspecific leakage of electrons and also driven by the activity of ETC complexes, mainly complex I and III. At complex I, generation of O2·− occurs primarily at the flavin mononucleotide prosthetic group, whereas ROS generation at complex III seems to occur at the ubisemiquinone which is bound at the Qo-site [57]. Minor amounts of ROS are generated by the other complexes or by reverse electron transport (RET). ETC-independent sources of mitochondrial ROS include monoamine oxidases (MAO) that generate H2O2 during deamination of different neurotransmitters, and NADPH oxidase 4 (NOX4) which produces O2·−. Production of ROS is counterbalanced by ROS detoxification to regulate ROS homeostasis, including a complex antioxidant system within mitochondria. Manganese superoxide dismutase (MnSOD) converts O2·− to H2O2, which can then be reduced to H2O by catalase and/or an antioxidative system comprised of gluthione, gluthathione peroxidases (Gpx), peroxiredoxins (Prx), and thioredoxins (Trx), dependent on the redox status and availability of reducing equivalents within mitochondria [5859]. Non-enzymatic antioxidant mechanisms include cytochrome c and coenzyme Q. Next to a physiologic function in modulating intracellular signaling, increased mitochondrial ROS induce oxidative damage to DNA, proteins and lipids, and may trigger a variety of pathological pathways involved in mitochondrial and cellular damage. Because of their short half-live, it is assumed that ROS predominantly cause harm close to their origin.
Good evidence exists for increased mitochondrial ROS in rodent and human DbCM [16336061]. Attenuation of mitochondrial ROS by overexpression of catalase, MnSOD or Prx3, or by treatment with mitochondria-targeted antioxidant agents such as Mito-TEMPOL, did attenuate mitochondrial oxidative stress, mitochondrial structural and functional defects, cardiac hypertrophy and contractile dysfunction, thereby establishing the causal link between mitochondrial ROS and development of DbCM [62636465]. Regarding sources and consequences of mitochondrial ROS, incubation of cardiomyocytes from diabetic OVE26 mice in high-glucose medium lead to increased ROS production, which was prevented by inhibition of complex I or II, along with improvement of cardiomyocyte contractility, whereas incubation of wildtype cardiomyocytes in high-glucose medium did not result in increased ROS generation. These results not only showed that hyperglycemia drives mitochondrial ROS production but also imply that certain diabetes-induced mitochondrial changes may predispose cardiac mitochondria of OVE26 mice to generate ROS [64]. Given that ETC defects can increase ROS generation by increasing the reductive state of the ETC, resulting in electron leakage, preexisting defects of the ETC could be one such predisposition for ROS production. Such defects in the ETC may result from direct protein damage, as has been exemplarily been shown for lipid peroxidation or protein tyrosine nitration of ETC complexes in STZ-diabetic rats [6066]. Insulin treatment removed lipid peroxidation of complex II and normalized respiration rates and complex II activity, suggesting ROS-induced protein damage in the ETC as a mechanism of mitochondrial dysfunction in these diabetic hearts. Similar to hyperglycemia, increased FAO may also increase ROS by increased electron delivery, but also by shifting fatty acyl-CoAs into pathways that mediate lipotoxicity, or induction of mitochondrial fission [455667].
Furthermore, db/db cardiomyocytes simultaneously subjected to energetic stress and redox stress by hyperglycemia and isoproterenol treatment resulted in an impaired transition from state 4 to state 3 respiration, which accounted for increased ROS generation both from forward and RET [68]. This was likely related to decreased levels of glutathione (GSH) and Trx2, and the resulting more oxidizing environment correlated with impaired excitation-contraction coupling. Resetting the mitochondrial redox balance by exogenous application of GSH or with palmitate, mediated by the TrxR2/Trx2/Prx3 system, normalized excitation-contraction coupling, which led the authors to propose that the inability of the diabetic heart to deal with an increase in energy demand may result from a perturbation of the mitochondrial redox status [68]. Another mechanism which may support impaired function of antioxidant enzymes is reduced levels of the mitochondrial deacetylase SIRT3, which promotes increased acetylation and thereby inactivation of MnSOD [69]. Other recently discovered mechanisms favoring mitochondrial ROS production and promoting DbCM might be increased levels of mitochondrial calpains or MAOs [6570]. Mimicking the increased mitochondrial levels of calpain 1 in diabetic hearts by targeted upregulation of calpain-1 in mitochondria induced cleavage of the ATP5A1 subunit of the FOF1-ATPase, and increased O2·− production and apoptosis in cardiomyocytes. Conversely, selective inhibition of mitochondrial calpains attenuated disruption of ATP synthase, decreased mitochondrial O2·− production, and prevented apoptosis in cardiomyocytes of STZ-diabetic mice, suggesting increased mitochondrial calpains as another mitochondrial mechanism contributing to DbCM [65]. Levels of MAO-A and MAO-B were also increased in the same model of DM, and increased H2O2 production was markedly attenuated by administration of MAO inhibitors [70].
Remodeling of the mitochondrial proteome
Transcriptional regulation is generally considered the mode of choice to adapt to chronic stimuli or diseases, and impaired expression of ETC subunits has been proposed as a cause of impaired mitochondrial function in numerous diseases, including cardiac pathologies. In failing hearts, a concerted downregulation of ETC subunits has been proposed to contribute to impaired mitochondrial function and energy depletion [56]. In the diabetic heart, a number of ETC subunits have been reported to be downregulated using immunoblotting of single proteins, although inconsistent results have been observed between studies and models [15]. More details were revealed by recent proteomics analyses of heart mitochondria, when even the proteome of the two distinct mitochondrial subpopulations has been analyzed. Subsarcolemmal mitochondria from db/db hearts showed several impairments, including a decrease in size and internal complexity, displayed decreased state 3 respiration and ATP synthesis rates, decreased ETC complex activities, and increased oxidative damage, whereas interfibrillar mitochondria were literally unaffected. Using isobaric tags for relative and absolute quantification (iTRAQ) and multi-dimensional protein identification technologies, a predominant decrease of protein subunits of the ETC, the ATP synthase, and of proteins of the mitochondrial protein import machinery was revealed, whereas these protein were nearly unaffected in interfibrillar mitochondria [71]. While such remodeling of the ETC may impair electron flow through the ETC and thereby overall ATP regeneration, such ETC defects are also assumed to increase the reduced state of the ETC, thereby facilitating electron leak and subsequent generation of superoxide.
Posttranslational modifications
Chronically increased protein O-linked beta-N-acetylglucosamine glycosylation (O-GlcNAcylation) induced by hyperglycemia contributes to cardiomyocyte dysfunction in DM. Several protein targets of O-GlcNAcylation have been identified in the diabetic heart, including phospholamban, calmodulin-dependent protein kinase II (CAMKII), actin, GATA binding protein 4 (GATA4), cytochrome c oxidase subunit 1 (COX1), dynamin-related protein 1 (DRP1), or forkhead box protein O1 (FOXO1), indicating a potential impact on various pathways whose impairment have been proposed to contribute to DbCM (for review see [72]). It has been shown that hyperglycemia leads to increased O-GlcNAcylation of CaMKII and promotes CaMKII-dependent Ca2+ release from the sarcoplasmic reticulum (SR), while removal of O-GlcNAcylation from myofilaments of cardiomyocytes in a mouse model of T1DM could restore Ca2+ sensitivity [7374]. O-GlcNAcylation might also facilitate hypertrophic signaling, partially transmitted by the activation of different transcription factors such as nuclear factor of activated T-cells (NFAT), GATA4, or myocyte enhancer factor 2C (MEF2C) [7576]. With regards to mitochondrial mechanisms, O-GlcNAcomic profiling found that over 88 mitochondrial proteins can be O-GlcNAcylated during inhibition of O-GlcNAcase, with the OXPHOS system as a major target. This O-GlcNAcylation was associated with increased mitochondrial oxygen consumption rates, ATP production rates, and an enhanced threshold for mitochondrial permeability transition pore (mPTP) opening, indicating that O-GlcNAcylation regulates mitochondrial biology [77]. In neonatal rat cardiomyocytes incubated in hyperglycemic medium, several subunits of the ETC, such as NDUFA9 of complex I, subunits core 1 and core 2 of complex III, and subunit I of complex IV have been found to be O-GlcNAcylated [78]. This O-GlcNAcylation was however associated with impaired activity of complex I, III, and IV, and removal of O-GlcNAcylation by overexpression of O-GlcNAcase normalized complex activities, besides a normalization of ATP levels. In another study though, a mislocalization of O-GlcNAc transferase (OGT) within mitochondria, leading to impaired interaction of OGT with complex IV, has been proposed to be responsible for impaired complex IV activity in diabetic hearts [79]. Thus, O-GlcNAcylation represents an important posttranslational modification that regulates mitochondrial function, although the functional consequences of increased O-GlcNAcylation in diabetic hearts remains to be elucidated in more detail.
Other modulators of posttranslational modifications within mitochondria are the protein family of SIRTs. SIRTs are nicotinamide adenine dinucleotide (NAD)+-dependent deacylases that are capable of removing a variety of different posttranslational modifications from protein lysine residues of a target protein, thereby regulating target protein function. SIRT3 is primarily localized within mitochondria and predominantly acts as a deacetylase, thereby counterbalancing the protein acetylation driven by acetyltransferases and non-enzymatic acetylation. Accordingly, lack of SIRT3 shifts the balance of acetylation towards increased protein acetylation, as was similarly observed in db/db hearts, including validated targets of SIRT3 such as LCAD and MnSOD [8081]. This increased protein acetylation in db/db hearts was associated with a decreased NAD+/NADH ratio and decreased expression and activity of SIRT3 [80]. Treatment with garlic or exogenous application of H2S restored the decreased NAD+/NADH ratio in db/db hearts and enhanced expression and activity of SIRT3, along with attenuation of hyperacetylation of ETC subunits and improvement of mitochondrial respiration and ATP synthesis [81]. In addition, H2S treatment normalized increased FAO rates and decreased pyruvate dehydrogenase (PDH) activity in db/db hearts, which improved respiratory function, ATP synthesis and ejection fraction, implying impaired SIRT3 activity in the pathogenesis of altered substrate utilization and impaired energetics in diabetic hearts. Accordingly, the authors proposed a model in which H2S administration improves the NAD+/NADH ratio and thus SIRT3 activity, leading to a myocardial substrate oxidation pattern that more closely resembles the physiologic myocardial substrate preference, thereby increasing the efficiency of ATP regeneration [80]. If true, it will be of interest to understand how cardiomyocytes will deal with the increased delivery of lipids within the cardiomyocytes in this setting, or if this normalization of the substrate oxidation pattern will in fact inhibit sarcolemmal FA uptake. In another study, cardiomyocyte apoptosis was increased and mitochondrial morphology (swelling, structural damage) was impaired in hearts of STZ-diabetic mice, accompanied by a suppression of autophagy, including mitophagy [82]. Prevention of a decrease in SIRT3 expression in neonatal mouse cardiomyocytes incubated in high glucose medium by SIRT3 overexpression restored autophagy and mitophagy and attenuated mitochondrial damage and apoptosis. In addition, overexpression of SIRT3 increased Parkin expression, suggesting that a suppression of SIRT3-Parkin signaling may have mediated the downregulation of mitophagy in diabetic hearts [82]. The potential role of the other mitochondrial SIRTs, SIRT4 and SIRT5, in the pathogenesis of DbCM and related mitochondrial defects remains to be elucidated.
Mitophagy and mitochondrial biogenesis
Autophagy represents a cellular mechanism by which damaged cellular components, including proteins, lipids, or cell organelles, are delivered to autophagosomes to ultimately degrade them following fusion with a lysosome. Autophagy has been shown to be crucial for the development, maturation and the function of the heart [83]. The specific targeting and removal of mitochondria by autophagy, i.e., mitophagy, is achieved by the pathways of phosphatase and tensin homolog-induced putative kinase 1 (PINK1) and the E3 ubiquitin ligase Parkin, and by many other proteins in the mitochondrial membrane or the cytosol [848586878889]. Recently, Tong et al. [90] showed that suppression of mitophagy either by deletion of autophagy-related protein kinase 7 (Atg7) or Parkin exacerbated DbCM in high-fat fed mice. Restoring mitophagy by injection of Tat-Beclin1 attenuated mitochondrial dysfunction, decreased lipid accumulation and protected against diastolic dysfunction [90]. In this study, high-fat diet upregulated mitophagy which highlights mitophagy as a protective compensatory mechanism in DbCM by which dysfunctional mitochondria can be dismissed. Together with the earlier made observations that PINK and Parkin levels are reduced in hearts of diabetic mice, one can speculate that either mitophagy is impaired in later stages of DM, or that the induction of mitophagy may not be sufficient to balance the mitochondrial damage caused by other mechanisms of DbCM [9192]. Another hypothesis would be that upregulated mitophagy is not adequately matched with subsequent mitochondrial biogenesis in diabetic hearts, thereby resulting in a decrease of total mitochondrial content.
Removal of mitochondria by mitophagy needs to be counterbalanced by mitochondrial biogenesis to maintain mitochondrial content of the cell. Mitochondrial biogenesis is thus part of the physiological turnover process of mitochondria that is predominantly regulated by PCG-1α signaling, which coactivates multiple transcription factors such as PPARα, estrogen receptor-related α (ERRα), nuclear respiratory factor 1 and 2 (NRF1/2), and mitochondrial transcriptionfactor A (mtTFA) [9394]. In patients with T2DM, mitochondrial biogenesis may be reduced in different organs, including the heart [95969798]. In contrast, myocardial PGC-1α expression is rather increased in animal models of DM, along with increased mitochondrial DNA content and increased mitochondrial area as well as count in the hearts of diabetic mice, both in models of T1DM and T2DM [3499100101102]. Shen et al. [64] showed that, in OVE26 mice, increased mitochondrial area and number were accompanied by increased mitochondrial damage and reduced respiratory control ratio of the mitochondria. These observations may imply that, in rodent models of DbCM, increased mitochondrial biogenesis may be the attempt to generate new and functionally intact mitochondria to compensate for mitochondrial damage and dysfunction in diabetic hearts. However, in combination with impaired mitophagy, this process of increased mitochondrial biogenesis may not be sufficient to remove all damaged mitochondria and/or to replace them by newly generated intact mitochondria. Also, the possibility exists that newly generated mitochondria are immediately damaged again by persistent factors that induce mitochondrial damage (e.g., oxidative stress), or that the process of actual biogenesis of mitochondria per se may be defective, resulting in generation of rather defective than functionally intact mitochondria. Further studies are needed to clarify the complex role of mitochondrial biogenesis in the diabetic heart, to address the question why mitochondrial biogenesis and/or the respective signaling may be different between human and rodent DbCM, and whether therapeutic induction of mitochondrial biogenesis, as is discussed for many other cardiac pathologies, may represent a way to cure mitochondrial dysfunction in DbCM [103].
Mitochondrial fission and fusion
Mitochondria are dynamic organelles that undergo continuous fusion and fission, a process that is required for mitochondrial biogenesis and that contributes to the regulation of mitochondrial energetics and ROS homeostasis. Mitochondrial dynamics are mediated by the action of a variety of distinct proteins, including the fission proteins Drp1 and fission 1 (Fis1), and the fusion proteins mitofusion 1 and 2 (Mfn1/2) and optic atrophy 1 (Opa1), among others. Mitochondrial fusion seems to overweigh during nutrient starvation or energy demanding states, leading to elongated tubular mitochondria, whereas fission promotes smaller fragmented mitochondria and seems to be induced by caloric excess [104]. Fission and fusion also have an impact on mitophagy where fission can separate damaged segments of mitochondria and induce mitophagy, whereas mitochondria appear to be degraded less likely following fusion [94105]. In the heart, deletion of either fusion or fission related proteins like Mfn1 and 2 or Drp 1 can lead to cardiomyopathy [106107]. With regards to DM, Yu et al. [108] showed that incubation of H9C2 rat cardiomyoblasts in high glucose medium induced a rapid mitochondrial fragmentation via Drp1 signaling which led to overproduction of ROS. In hearts of T1DM rodents though, fragmentation of mitochondria rather occurs as a response to chronic hyperglycemia. While mitochondrial morphology was unaltered after 3 weeks of hyperglycemia, electron microscopy revealed distorted vacuous mitochondria with decreased matrix electron density after 5 weeks of hyperglycemia, accompanied by increased proteolytic cleavage of Opa1 leading to more fragmented mitochondria due to impaired fusion [109]. Of interest, ROS production was unaffected after 3 weeks of hyperglycemia, whereas after 5 weeks following hyperglycemia, dysfunction of the ETC complexes and increased ROS production (primarily from RET) were apparent in these hearts [109]. These observations rather suggest a progressive impairment in mitochondrial dynamics in DbCM that may be caused by a chronic dysbalance of fusion and fission. In addition, given that mitochondrial fission can increase ROS production and vice versa, a vicious cycle between ROS and mitochondrial fragmentation may develop during chronic hyperglycemia, with one mechanism being able to trigger and further impair the other. Support for a dysregulated balance of fusion and fission has also been demonstrated in a study of Makino et al. [110] who showed decreased levels of Opa1 in mouse coronary endothelial cells isolated from diabetic mice while Drp1 levels were also increased. Of further interest, lowering of oxidative stress by TEMPOL treatment restored normal mitochondrial morphology, suggesting that a chronic increase in ROS may trigger mitochondrial fragmentation in DbCM, and that ROS scavenging may be an effective way to interrupt the vicious cycle of increased ROS and impaired mitochondrial dynamics.
Besides changes in expression, posttranslational modifications such as increased O-GlcNAcylation of Opa1 and Drp1 have also been shown to contribute to the changes in mitochondrial dynamics in the diabetic milieu. High glucose levels increase O-GlcNAcylation of Opa1, and reducing this modification attenuates mitochondrial dysfunction [111]. O-GlcNAcylation of Drp1 is observed in hearts of T2DM mice, which decreases phosphorylation of Drp1 and thereby induces translocation of Drp1 onto mitochondria and eventual fragmentation [112]. Insights into upstream mechanisms of impaired mitochondrial dynamics in DbCM come from observations in mice overexpressing long-chain acyl-CoA synthetase 1 (ACSL1) as a model of cardiac lipotoxicity. In this model, reduced phosphorylation of Drp1 and altered proteolytic processing of Opa1 led to increased mitochondrial fission, suggesting that increased lipid uptake, as typically observed in diabetic hearts, may trigger mitochondrial fragmentation and thus mitochondrial dysfunction and ROS [45]. The link between FA metabolism and mitochondrial dynamics was also endorsed by Kolleritsch et al. [113] who could show that cardiomyocyte-specific overexpression of mutated perilipin 5 resulted in reduced cardiac lipolysis and attenuation of mitochondrial fission, accompanied by less mitochondrial recruitment of Drp1 and decreased phosphorylation of the Drp1 interaction partner, mitochondrial fission factor. Of note, a recent study by Hu et al. [114] not only demonstrated that mitochondrial fission is impaired in T2DM db/db hearts, but also that the proposed mechanism of reduced Mfn2 expression may have been a consequence of reduced expression and binding of PPARα to the Mfn2 promoter, thus providing more evidence of a link between lipid metabolism and impaired mitochondrial dynamics in DbCM. According to these data, the authors proposed a model in which a decrease in PPARα expression in the rather chronic DM situation would impair expression of PPARα and Mfn2, thereby inducing mitochondrial fission which results in mitochondrial respiratory dysfunction, increased mitochondrial ROS generation and mitochondria-dependent apoptosis [114].
Mitochondrial Ca2+ handling
Ca2+ is the pivotal messenger for excitation-contraction coupling in the heart which connects the electrical stimulus to the contraction of the myocytes. During cardiomyocyte depolarization, a small Ca2+ influx via sarcolemmal L-type Ca2+ channels (LTCC) triggers opening of the ryanodine receptor (RyR) of the SR, resulting in a large release of Ca2+ from the SR into the cytosol, thereby activating myofilament cross-bridge formation and triggering cardiomyocyte contraction [115]. To end the contraction cycle, Ca2+ is mainly removed from the cytosol by reuptake into the SR via the sarco/ER Ca2+-ATPase 2a (SERCA2a), and a smaller amount is removed into the extracellular space via the Na+/Ca2+ exchanger [115]. To match increased energy demand with energy production, cytosolic Ca2+ transients also trigger mitochondrial Ca2+-uptake, facilitated by the mitochondrial calcium uniporter (MCU). In mitochondria, Ca2+ can stimulate ATP regeneration through enhancing the activity of ETC complexes I, II, and IV, and by activating PDH, α-ketoglutarate dehydrogenase, and isocitrate dehydrogenase. Furthermore, the FOF1-ATPase requires Ca2+ for its activity, and it has been estimated that this activation may have been responsible for more than 60% of the Ca2+-induced activation of OXPHOS, whereas the contribution of Ca2+-sensitive dehydrogenases may have accounted for only 40% [116].
While there is evidence that cellular Ca2+ handling is impaired in DbCM due to decreased SERCA2a expression, reduced activity of RyR or decreased LTCC expression, literally no data on mitochondrial Ca2+ handling in DbCM have been published [117118]. However, studies showed that myocardial mitochondrial Ca2+ uptake was reduced in STZ-induced T1DM rats and in T2DM ob/ob mice [119120]. Recently, Ji et al. [121] found that cardiomyocyte expression of mitochondrial calcium uptake protein 1 (MICU1), a regulatory subunit of the MCU, was downregulated in db/db mice at 12 weeks of age, accompanied by mitochondria-dependent intrinsic apoptosis. Reconstitution of MICU1 normalized cardiac function, attenuated cardiac hypertrophy and fibrosis, and inhibited apoptosis in this mouse model [121]. In addition, increased mitochondrial Ca2+ uptake through upregulation of MICU1 attenuated mitochondrial ROS and ROS-triggered apoptosis [121]. In another study, expression of MICU1 was increased in hearts of STZ-diabetic mice, however levels of MCU and essential MCU regulator (EMRE; subunit of MCU) were decreased, thus also leading to a decrease in mitochondrial Ca2+ uptake, mitochondrial function, and cardiac function. Restoring MCU expression also rescued cardiac and mitochondrial respiratory dysfunction, underscoring the proposal of mitochondrial Ca2+ restoration as a potential target for therapeutic intervention [122123]. One has to keep in mind though that triggering of mitochondrial permeability transition due to Ca2+ overload may complicate this therapeutic strategy [124125]. Of note, increasing Ca2+ uptake by overexpression of MICU1 may be beneficial in diabetic hearts, but resulted in increased mortality in non-diabetic mice [121].
Dysregulation of microRNAs
MicroRNAs (miRNAs) are single-stranded non-coding RNA molecules that are about 22 nucleotides long and that regulate protein levels by inhibition of mRNA translation or degradation of mRNA [126]. Dysregulation of a large number of different miRNAs has been implicated by now to contribute to the pathogenesis of cardiovascular diseases, including HF and DbCM, as reviewed elsewhere [127128]. For example, miR133a expression was reduced in STZ-treated diabetic mice displaying ventricular dysfunction and hypertrophy, and transfection of miR133a mimics to cardiomyocytes incubated in high glucose medium prevented hypertrophic changes [129]. Also, miR-30c expression was decreased in diabetic mice with cardiomyocyte hypertrophy and in humans with DbCM, and overexpression of miR-30c attenuated hypertrophy in cardiomyocytes treated with glucose, possibly via decreased expression of cell division control protein 42 homolog (Cdc42) and p21activated kinase 1 (Pak1) [130]. Interestingly, it has been shown that not only nuclear-encoded proteins but also mitochondria-encoded proteins can be regulated by miRNAs [131132]. With regards to DbCM, a number of studies suggest that mitochondrial dysfunction may also result to some part from alterations in miRNA action. Overexpression of miRNA-195 observed in T1DM and T2DM hearts may contribute to downregulation of SIRT1 in these hearts, and impaired SIRT1 activity can then be assumed to downregulate oxidative metabolism, mitochondrial function and to increase ROS production [133]. PGC-1α is a direct target of miRNA-29a, and myocardial levels of miRNA-29a were shown to be decreased in STZ-induced diabetic animals, a mechanism that may contribute to induction of PPARα and FAO, as well as to mitochondrial biogenesis [134]. Baseler et al. [135] found increased levels of miRNA-141 in T1DM mice, which may impair the activity of solute carrier family 25 member 3 (Slc25a3) and thus import of inorganic phosphate and subsequent ATP regeneration. Also, an increased level of miRNA-378 in interfibrillar cardiac mitochondria of STZ-diabetic mice has been shown to impair the translation of the ATP6 subunit of the FOF1-ATPase [136]. These studies imply that miRNAs may interfere with different pathways, proteins and enzymes that are essential to maintain oxidative function of mitochondria. It is thus quite plausible that miRNA dysregulation contributes to mitochondrial dysfunction in DbCM, although the elucidation of exact contributions, the complexity of interactions, and the further characterization of all mitochondria-regulating miRNAs will be a major task of future studies.
POTENTIAL THERAPEUTIC STRATEGIES TARGETING MITOCHONDRIA IN DbCM
Both our increased understanding of mitochondrial mechanisms contributing to DbCM and the increasing elucidation of mechanisms of action of antidiabetic drugs has increased the attention for mitochondria as a therapeutic target in diabetic heart disease. A number of antidiabetic medications currently used in clinical practice may already directly or indirectly attenuate mitochondrial defects associated with DbCM, including metformin. ER stress is a typical trait in DbCM, and a recent study demonstrated that induction of ER stress by thapsigargin treatment in non-diabetic mice impairs mitochondrial respiration, may induce mPTP opening, and increases mitochondrial oxidative stress [137]. Additional treatment of these mice with metformin normalized all of the mitochondrial abnormalities, potentially by activation of AMP activated protein kinase, suggesting that metformin may cure mitochondrial defects in DbCM induced by ER stress or even by other causes.
Mostly discussed these days though are the potential mechanisms by which sodium glucose cotransporter 2 inhibitors (SGLT2i) improve cardiovascular outcomes in diabetic subjects. Results of several phase 3 clinical trials demonstrated that additional treatment of diabetic subjects with SGLT2i leads to a significant reduction in cardiovascular endpoints, including the classical major adverse cardiovascular event (MACE) endpoint (non-fatal myocardial infarction, non-fatal stroke, cardiovascular death), all-cause mortality, and/or in particular, hospitalization for HF [138139140]. Although each trial investigating outcomes of specific SGLT2i (empagliflozin, canagliflozin, dapagliflozin) showed slight differences in outcomes and patient inclusion criteria (patients with cardiovascular risk factors vs. patients with known cardiovascular disease), the effects of SGLT2i are generally considered rather a class effect. Accordingly, the European Society of Cardiology recently adapted the guidelines on the treatment of diabetic patients based on this new clinical trial evidence, now suggesting SGLT2i even as a first line therapy in diabetic subjects with high or very high cardiovascular risk or known cardiovascular disease [141]. Not included in the new guidelines yet are the results of the recent Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction (DAPA-HF) trial, which demonstrated that dapagliflozin treatment lead to a reduction of the composite endpoint of worsening HF or cardiovascular death in patients with HF with reduced ejection fraction, irrespective whether the patients suffered from diabetes or not [142]. Given the therapeutic efficacy of dapagliflozin in non-diabetic patients, the question rises in how far glucose lowering indeed contributes to the beneficial effects on macrovascular outcomes, mortality, and HF worsening. Furthermore, this trial indicates that SGLT2i treatment may target myocardial mechanisms underlying systolic HF per se, some of which are also present in patients with DbCM, including mitochondrial defects.
Despite the likely absence of SGLT2 in the myocardium, direct myocardial mechanisms of SGLT2i have been identified or proposed that may affect mitochondrial function. The cardiac Na+/H+-exchanger 1 (NHE1) has been identified as a target of SGLT2i. In isolated cardiomyocytes from rabbits and mice, treatment with empagliflozin inhibited NHE1 flux, reduced cytosolic Na+ and Ca2+ levels, and increased mitochondrial Ca2+ levels, likely by direct binding of empagliflozin to NHE1 [143144]. SGLT2i may thereby attenuate defects in both cytosolic and mitochondrial Ca2+ handling and may increase ATP regeneration by activating mitochondrial Ca2+-sensitive dehydrogenases [143144]. Another mechanism may be related to SGLT2i-mediated alterations in myocardial ketone metabolism. SGLT2i treatment results in increased serum β-hydroxybutyrate levels which led to the “thrifty substrate” hypothesis, proposing that mild hyperketonemia and a subsequent relative increase in myocardial oxidation of ketone bodies may increase cardiac work per oxygen consumed, i.e., cardiac efficiency [144]. Indeed, continuous treatment with empagliflozin resulted in a reduction of glucose oxidation, in an increase in myocardial uptake of ketone bodies, FA, and branched-chain amino acids (BCAA), and in enhanced activity and/or expression of enzymes involved in ketone body, FA and BCAA metabolism in a porcine model of HF [145]. While this substrate switch was accompanied by increased systolic function and attenuation of left ventricular remodeling, it needs to be kept in mind that the animals were not diabetic and a similar effect needs to be demonstrated in diabetic animals as well [145]. Of note, subjecting pigs to myocardial ischemia reperfusion combined with short term pretreatment with canagliflozin preserved cardiac function and efficiency during ischemia but had no effect on the pattern of myocardial substrate uptake, including ketone bodies, potentially arguing against the “thrifty substrate” hypothesis, although energy metabolism during ischemia and reperfusion markedly differs from substrate utilization in chronic HF or DbCM [146].
Similar to SGLT2i, some glucagon-like peptide 1 receptor agonists (GLP1RA) have demonstrated a reduction of cardiovascular endpoints in clinical trials, including MACE or CV death alone, thus resulting in a similar guideline recommendation as described before for SGLT2i [147148]. Although only few data are available on the underlying mechanisms of GLP1RA, mitochondrial mechanisms may also be affected by GLP1RA treatment. In a rat model of chronic hypoxia, treatment with liraglutide attenuated activation of the mitochondrial apoptosis pathway, increased activation of protective mitophagy via increased expression of Parkin, attenuated mitochondrial oxidative stress, and prevented energy depletion [149].
Besides positive effects of antidiabetic drugs on mitochondrial biology in DbCM, other approaches may also be promising therapeutic strategies to improve mitochondrial function in DbCM. Given the multiple detrimental effects of mitochondrial oxidative stress in DbCM, mitochondrial ROS scavenging is an obvious potential treatment strategy in DbCM. Several mitochondria-targeted agents have demonstrated the capacity to attenuate mitochondrial oxidative stress in preclinical studies, such as MitoQ, MnTBAP, or MitoTempol. Treatment with MnTBAP reversed cardiac mitochondrial oxidative stress and improved mitochondrial bioenergetics in a mouse model of the metabolic syndrome [150]. In leukocytes of T2DM patients, treatment with MitoQ attenuated mitochondrial ROS production and showed antiinflammatory and antioxidant effects [151].While MitoQ has shown beneficial effects in different cardiac pathologies as reviewed elsewhere, studies evaluating antioxidative treatment in DbCM are however lacking [8152]. Another approach to attenuate mitochondrial oxidative damage is supplementation with tetrahydropterin (BH4), which is required for full functional activity of endothelial nitric oxidase synthase (eNOS). In diabetes, BH4 is oxidized to BH2, and low BH4 levels are known to result in decreased eNOS activity and in increased superoxide production by eNOS. Given the proximity of eNOS to the outer mitochondrial membrane, such eNOS-derived ROS may cause mitochondrial damage. Therapeutic application of sepiapterin, a BH4 precursor, together with L-citrulline as an L-arginine precursor, prevented cardiac dysfunction in diabetic db/db mice and also attenuated myocardial infarct size in this rodent model [153]. To date, none of the above discussed antioxidants have been investigated in human trials for potential beneficial effects in DbCM or HF.
Some beneficial effect in DbCM may also be achieved by modulation of the myocardial (mitochondrial) substrate oxidation pattern towards a physiological substrate utilization pattern. For example, the anti-anginal agent trimetazidine inhibits long-chain 3-ketoacyl coenzyme A thiolase in the β-oxidation spiral and thereby reduces FAO, improves systolic function and exercise tolerance, and reduces N-terminal-pro hormone brain natriuretic peptide (NT-proBNP) levels in diabetic subjects with idiopathic dilated cardiomyopathy after 6 months of treatment when compared to placebo [154]. Furthermore, trimetazidine added to standard medical therapy showed beneficial effects on left ventricular ejection fraction in diabetic subjects with ischemic heart disease compared to placebo [155]. Ranolazine also inhibits FAO and activates PDH and has been shown to improve hemodynamics in HFpEF patients [156157]. In the Metabolic Efficiency with Ranolazine for Less Ischemia in Non-ST-Elevation Acute Coronary Syndromes trial (MERLIN-TIMI36) trial, ranolazine treatment reduced recurrent myocardial ischemia in diabetic patients [158]. Inhibition of CPT1 by perhexiline reduces FAO and has been shown to improve maximal oxygen uptake, to increase the phosphocreatine to creatine (PCr/Cr) ratio, improve left ventricular systolic function, as well as skeletal muscle energetics in chronic HF and cardiac energetics in patients with dilated cardiomyopathy [159160161162]. Data of ranolazine in patients with diabetic heart disease are lacking to date. Additional and larger trials investigating patients with DbCM with and without systolic HF are thus necessary to further evaluate a potential beneficial effect of metabolic modulation in diabetic subjects.
Finally, NAD+ levels are depleted in DbCM, possibly due to hyperactivation of the NAD+ consuming DNA repair enzyme, poly (ADP-ribose)-polymerase 1 (PARP-1). Since NAD+ is an essential cosubstrate for SIRTs, cellular NAD+ depletion may impair the activity of both intramitochondrial and extramitochondrial SIRTs [43163]. A recently published study showed that chronic oral application of the NAD+ precursor nicotinamide riboside (NR) is well tolerated and able to elevate NAD+ levels in healthy adults, thereby opening a new treatment option for several cardiac diseases associated with decreased NAD+/NADH ratios, including DbCM [164]. In preclinical studies, activation of SIRTs by restoration of NAD+ levels by treatment with NAD precursors (nicotinamide mononucleotide or NR) or by overexpression of nicotinamide phosphoribosyltransferase improved cardiac function in different models of HF or following ischemia reperfusion [165166]. These effects have been proposed to be mediated by activation of SIRT1 and/or SIRT3. Pharmacological inhibition of PARP-1 would represent another approach to maintain cellular NAD+ levels. Indeed, application of the PARP-1 inhibitor INO1001 attenuated oxidative stress, inflammation, fibrosis, and increased expression of PGC-1α in hearts of T2DM mice via activation of SIRT1 [167].
CONCLUSIONS
In the current review, we discussed mechanisms and pathways that are disturbed in the diabetic heart and that render mitochondria susceptible to damage and dysfunction. These mitochondrial defects contribute to many pathologic features of the diabetic heart, including impaired contractility, which puts mitochondria at center stage of the pathophysiology of DbCM. The next step has to be a comprehensive confirmation of many of these mechanisms in the human diabetic heart to gain more insight if potential treatment targets identified in animal models may also be a valid target in humans. Such data are instrumental for the development of potential therapies to prevent or at least attenuate diabetes-related mitochondrial defects in the heart and to increase the chance that the therapeutic strategies also prove effective in clinical trials. The significance of improving myocardial mitochondrial abnormalities in diabetic subjects is emphasized by the fact that some beneficial effects of cardioprotective antidiabetic drugs (e.g., SGLT2i, metformin) seem to be mediated by direct or indirect effects on mitochondria. With increasing understanding of mitochondrial biology, the increasing use of large animal models, and by using rapidly evolving new scientific technologies, mitochondrial medicine may become a realistic therapeutic option in the closer future. Given the widespread involvement of mitochondrial defects in human disease, even including the pathogenesis of DM itself, the successful development of mitochondrial therapies will likely benefit numerous patients suffering from many other diseases beyond DbCM as well.
 XML Download
XML Download