

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Ischemic and traumatic brain injuries are the major acute central nervous system disorders that need to be adequately diagnosed and treated. Studies on the biomarkers for these disorders have been executed [12]. But more research is needed for the development of useful biomarkers for these disorders.
Specialized pro-resolving mediators (SPMs) are a group of bioactive lipids involved mainly in the resolution of inflammation. They include lipoxin A4 (LXA4) derived from arachidonic acid, resolvin Es (RvEs) from eicosapentaenoic acid (EPA), resolvin Ds (RvDs) from docosahexaenoic acid (DHA), and others. The changes in the plasma SPMs levels have been reported in various diseases, including pneumonia [3], preeclamsia [4], arthritis [5], the carotid disease [6], type 2 diabetes [7], and chronic spontaneous urticaria [8]. However, there have been no reports on the changes in the plasma SPMs levels after acute brain injuries. Thus, we examined the changes in the plasma levels of some SPMs (LXA4, RvE1, RvE2, RvD1, and RvD2) after acute brain injuries, i.e., global cerebral ischemic (GCI) and traumatic brain injuries (TBI) in the present study.
Additionally, plasma levels of CD59, a complement regulatory protein [9], were reported to be increased in acute myocardial infarction patients [10]. However, there have been no reports on the changes in the plasma CD59 levels after acute brain injuries. Thus, we also examined the changes in the plasma CD59 levels after GCI and TBI.
METHODS
Global cerebral ischemia induction
GCI was induced by bilateral common carotid artery occlusion as previously described [1718]. Rats were deeply anesthetized with isoflurane (1%–2% for maintenance; 3% for induction) in a 70:30 mixture of nitrous oxide/oxygen using a isoflurane vaporizer (VetEquip, Livermore, CA, USA) and core temperature was kept at 36.5℃–37.5℃ with a homeothermic blanket control unit (Harvard Apparatus, Holliston, MA, USA). A catheter filled with heparin was inserted into the femoral artery. Cannulation was used for observation of arterial blood pressure and draining blood. Both common carotid arteries were exposed through a midline neck incision and loosely encircled with a 4/O silk suture before the start of the occlusion. Electroencephalographic (EEG) probes were placed in bilateral burr holes. Systemic mean arterial pressure (MAP) was decreased within the range of 40 ± 10 mmHg by draining blood (7–10 ml) from the femoral artery. The exposed bilateral common carotid arteries were clamped when they were within the 40 ± 10 mmHg MAP range. Successful induction of GCI was confirmed based on isoelectricity using an EEG monitor. After observing the isoelectric EEG signal for 7 min, blood circulation was reinstated by unclamping the occlusion. Vitals and EEG signals were monitored until they returned to baseline. Following suture of the skin incision, anesthesia was discontinued. When rats showed spontaneous respiration, they were returned to a temperature-controlled incubator maintained at 37℃. Sham-operated rats received the same neck skin incision under isoflurane anesthesia, but common carotid artery occlusion was not performed.
Controlled cortical impact model for TBI
A controlled cortical impact model of experimental TBI was performed as previously described [1920]. Briefly, rats were anesthetized with isoflurane (1%–2% for maintenance; 3% for induction) in a 70:30 mixture of nitrous oxide and oxygen using an isoflurane vaporizer (VetEquip), and positioned in a stereotaxic apparatus (David Kopf Instruments, Tujunga, CA, USA). A craniotomy was made approximately 5 mm over the right hemisphere using a portable drill (2.8 mm lateral to the midline and 3 mm posterior to the bregma). A 3 mm flat-tip impactor was accelerated down to a 3 mm depth at a velocity of 5 m/sec using a controlled cortical impact device (Leica Impact One; Leica Biosystems, Nussloch, Germany). All rats were maintained at a core temperature of 36℃–37.5℃ with a homeothermic blanket control unit (Harvard Apparatus) during and after surgery, until ambulatory. Sham-operated rats only received craniotomy.
Assay of LXA4, RvE1, RvE2, RvD1, RvD2, CD59, and IL-6
After anesthesia, blood (1.5 ml) was collected from retro-orbital venous plexus of sham and experimental groups at 6, 24, 72, and 168 h after injury. The number of animals was 5 for sham and experimental groups. Plasma LXA4, RvE1, RvE2, RvD1, RvD2, CD59, and IL-6 levels were measured with ELISA (MyBioSource, San Diego, CA, USA). For normal plasma levels, blood was collected from three naïve animals.
Tissue preparation
Rats were deeply anesthetized with urethane (1.5 g/kg, intraperitoneal) in saline (0.9% NaCl) at a volume of 0.01 ml/g body weight. A toe pinch was used to evaluate the effectiveness of anesthesia. Rats were intracardially perfused with saline, followed by 4% paraformaldehyde in phosphate-buffered saline (PBS). The brains were post-fixed with 4% paraformaldehyde for 1 h and then immersed in 30% sucrose for cryoprotection. Thereafter, the entire brain was frozen and coronally sectioned on a cryostat microtome (CM1850; Leica, Wetzlar, Germany) at 30 µm thickness.
Assessment of neuronal death
Neuronal death was evaluated 3 days after GCI or TBI. To identify degenerating neurons, Fluoro-Jade B (FJB; Histo-Chem, Jefferson, AR, USA) staining was used as previously described [21]. The sections were photographed using a confocal microscope (LSM 710; Carl Zeiss, Oberkochen, Germany) with excitation and emission wavelengths of 480 nm and 525 nm, respectively.
Immunostaining
The sections were incubated with primary antibodies in PBS containing 0.3% Triton X-100 at 4℃ overnight as follows: goat anti-GFAP (diluted 1:1,000; Abcam, Cambridge, UK) or rabbit anti-Iba-1 (ionized calcium binding adaptor molecule-1; diluted 1:500, Abcam). After washing in PBS, fluorescent-conjugated secondary antibodies (diluted 1:250, Invitrogen, Carlsbad, CA, USA) were applied to detect GFAP or Iba-1 antibody for 2 h at room temperature. Sections were counterstained with DAPI (4,6-diamidino-2-phenylindole; diluted 1:1,000; Invitrogen). Fluorescence-stained sections were mounted on gelatin-coated slides and cover-slipped with DPX (Sigma-Aldrich, St Louis, MO, USA). The immunoreactions were observed under a confocal microscope (LSM 710; Carl Zeiss).
RESULTS
To examine the possible role of LXA4 as a biomarker of GCI and TBI, we measured the plasma LXA4 levels after GCI or TBI in rats (Fig. 1). As shown in Fig. 1A, the plasma LXA4 levels did not change up to 6 h but, tended to increase at 24 and 72 h, and to remain elevated until 168 h post-GCI (Fig. 1A). However, the changes did not reach statistical significance due to the slight shortage of the number of animals used (n = 5). Oppositely to the case of GCI, plasma LXA4 levels showed a tendency of decrease after TBI throughout the observation period (Fig. 1B).
Next, we examined the plasma RvE1, RvE2, RvD1 and RvD2 levels after GCI or TBI in rats (Fig. 2). The changes in the plasma RvE1, RvE2, RvD1, and RvD2 levels showed a pattern different from that of LXA4 after GCI. As shown in Fig. 2A, 2E and 2G, plasma RvE1, RvD1 and RvD2 levels showed a biphasic response to GCI; they significantly decreased at 6 h, but returned to the levels of the sham group at 24 h, and again decreased at 72 h after GCI. In contrast to the case of GCI, plasma RvE1, RvE2, RvD1 and RvD2 levels did not show significant changes after TBI (Fig. 2B, D, F, and H). Notably, in the GCI sham groups, the plasma resolvins levels remained decreased from 24 h up to 168 h, when compared with those at 6 h (Fig. 2), suggesting that the surgical procedure for the sham group itself decreases the plasma resolvins levels from a certain time-point after the procedure.
Next, we examined the plasma CD59 levels after GCI or TBI in rats (Fig. 3). As shown in Fig. 3A, plasma CD59 levels increased at 6 and 24 h, and returned to the levels of the sham group at 72 h post-GCI. However, plasma CD59 levels showed no changes after TBI (Fig. 3B).
Finally, we examined the plasma IL-6 levels after GCI or TBI in rats (Fig. 4). Plasma IL-6 levels significantly increased at 72 h post-GCI and the increase was maintained up to 168 h (Fig. 4A). The increase in the plasma IL-6 levels after TBI was more delayed; they were not changed up to 72 h, but markedly increased at 168 h (Fig. 4B).
GCI or TBI lead to considerable neuronal death in the hippocampus. In addition, previous studies suggested that activation of glial cells, such as microglia and astrocytes, reaches peak levels at 1 week after GCI or TBI [2223]. We examined neuronal death and glial activation after GCI or TBI in our models. The number of FJB+ neurons was remarkably increased 3 days after GCI or TBI, compared to sham-operated group (Fig. 5A). GCI or TBI promoted microglial activation, as evidenced by transition from a ramified to an amoeboid shape (Fig. 5B), and pathological changes of astrocytes, such as glial scar formation and reactive gliosis (Fig. 5C).
DISCUSSION
We observed a tendency of increase in plasma LXA4 levels after GCI (Fig. 1A), while plasma LXA4 levels tended to decrease after TBI (Fig. 1B). The increase in plasma LXA4 levels was reported in preeclampsia [4], whereas the decrease was reported in the cases of pneumonia [3] and chronic spontaneous urticaria [8]. The mechanisms for differing plasma LXA4 changes in different disorders remain to be studied.
In the mouse intestine ischemia reperfusion injury, intestinal tissue LXA4 levels are increased [24]. Notably, in the mouse brain ischemia reperfusion injury, LXA4 levels increase up to 8 h postinjury in the injured brain area [25]. The tendency of increased post-GCI plasma LXA4 levels (Fig. 1A) could be resulted from ischemia reperfusion-injured brain tissue.
In contrast to the plasma LXA4 levels, plasma RvE1, RvD1 and RvD2 levels decreased after GCI (Fig. 2A, E, G). The reason for the differential effects of GCI on LXA4 versus RvE1, RvD1 and RvD2 is not clear at the moment. However, it could be, at least in part, due to the different fatty acids as substrates for the synthesis of LXA4 and resolvins; LXA4 is derived from arachidonic acid, whereas RvE1 and RvE2 are from EPA, and RvD1 and RvD2 are from DHA. It is also to be noted that the earliest time point of changes is 24 h for LXA4, and 6 h for RvE1, RvD1 and RvD2 after GCI, suggesting that RvE1, RvD1, and RvD2 are more valuable for the early diagnosis of GCI.
Decease in the plasma resolvin levels was reported in baboon pneumonia [3] and in patients with acutely symptomatic carotid disease [6], whereas in patients with arthritis the plasma resolvin levels were increased [5]. In the present study, we found a biphasic decrease in the plasma RvE1, RvD1 and RvD2 levels after GCI (Fig. 2A, E, G). The mechanisms underlying this biphasic response remain to be identified.
As plasma CD59 levels were reported to be increased in acute myocardial infarction patients [10], our finding of increased plasma CD59 levels after GCI but not TBI (Fig. 3A, B) suggests that the increase in plasma CD59 levels indicates ischemic injuries of organs, such as heart and brain. It is to be noted that the plasma CD59 levels significantly increased as early as 6 h post-GCI up to 24 h post-GCI, suggesting its usefulness for the early diagnosis of GCI.
In consistence with previous reports [111213141516], plasma IL-6 levels increased after both GCI and TBI (Fig. 4). It is suggested that a common mechanism underlies the increase in plasma IL-6 between GCI and TBI. Notably, the increases in plasma IL-6 levels were rather delayed, i.e., at 72 h through 168 h post-GCI and at 168 h post-TBI, suggesting its usefulness for the diagnosis of GCI and TBI at a later time point.
Limitations of the present study are as follows. Correlations between the magnitude of the changes in the plasma SPMs and CD59 levels and the extent of the injury as well as the extent of recovery from the injury were not examined. Those correlations would be useful for monitoring disease progression and for validating therapy for injury. The earliest time point we used was 6 h. As plasma resolvins and CD59 levels significantly changed already at 6 h after GCI (Figs. 2, 3), the earlier time points need to be studied to obtain a more precise time-course. Blood samples were obtained from the naïve animals only at one time-point. As multiple blood samples were taken at sequential time-points from the sham and the experimental animals, the possible effects of the multiple sequential blood sampling itself on the decrease in the resolvins levels in the sham group (Fig. 2) could have been monitored in the naïve animals.
In conclusion, we firstly found some substances in plasma that differentially changes after acute brain injuries, i.e., GCI and TBI. Plasma LXA4 levels tended to increase after GCI, but to decrease after TBI. Plasma RvE1, RvD1 and RvD2 levels decreased after GCI, but not after TBI. Plasma CD59 levels increased after GCI, but not after TBI. These differential changes could be helpful for the differential diagnosis of GCI and TBI. Further research is needed to evaluate these differential changes after GCI and TBI for the useful biomarkers of these acute brain injuries.
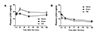

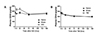


 XML Download
XML Download