

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
SHP2 is a non-receptor protein tyrosine phosphatase encoded by the PTPN11 gene which is located on chromosome 12q in humans [1]. The human PTPN11 gene was first cloned from a human umbilical cord cDNA library and originally named PTP2C [2]. SHP2 contains two SH2 domains (an N-terminal and a C-terminal SH2 domain) at its N-terminus, which are followed by a protein tyrosine phosphatase (PTP) domain [2]. Shortly after cloning, the crystal structure of SHP2 was revealed [3]. In the basal state, the N-terminal SH2 domain interacts with the PTP domain and prevents the catalytic domain from accessing the substrate [3]. Binding of the N-terminal SH2 domain (N-SH2) to a phosphopeptide triggers a conformational change in SHP2 and releases auto-inhibition by N-SH2, suggesting that the N-SH2 functions as a molecular switch [34]. Phosphorylated receptor tyrosine kinases and adaptor proteins, such as GRB2, serve as upstream activators for SHP2 [56]. SHP2 harbors two tyrosine phosphorylation sites (Y542 and Y580) of which phosphorylation can affect its activity upon stimulation with several growth factors [7]. Activation mechanisms of SHP2 might differ in different tissue and cell types, which remain to be further investigated. SHP2 is required for the full activation of the RAS-MAPK signaling pathway [568]. Either inhibiting activity or expression of SHP2 was shown to reduce RAS-MAPK activity in response to some, but not all, growth factors [910]. Accordingly, loss-of-function SHP2 mutations (Y279C, A461T, T468M, G464A) found in LEOPARD syndrome showed reduced catalytic activity as well as reduced MAPK activation compared to those of wild type SHP2 in response to EGF treatment in HEK293T cell [10]. Consistently, heart lysates from knock-in mice expressing a LEOPARD syndrome associated SHP2 Y297C mutation showed reduced ERK activation [11]. However, it is puzzling that MAPK activation was found to be increased in both a patient-derived iPSC (SHP2 T468M) and a zebrafish model (SHP2 A462T) of LEOPARD syndrome [1213]. The reason for the discrepancy between in vitro and in vivo experiments is not clear [14]. In addition to the RAS pathway, SHP2 has also been shown to activate the PI3K-AKT pathway, while it inhibits the JAK-STAT pathway [5151617]. The detailed molecular roles of SHP2 in signaling cascades have been extensively reviewed elsewhere [56].
Mutations in PTPN11 are associated with developmental disorders including Noonan syndrome (NS) and LEOPARD syndrome [1018]. Gain-of-function mutations in PTPN11 are associated with NS, while loss-of-function mutations are associated with LEOPARD syndrome [5610]. Interestingly, there are many overlapping features between NS and LEOPARD syndrome including developmental delays and learning difficulties [14]. It is not clear how the mutations resulting in opposite effects on the same signaling pathway can cause a similar phenotype. However, it is not unique to PTPN11 mutations that both overexpression and suppression of a particular gene result in the overlapping phenotypes by disrupting the fine-tuned regulation of the corresponding signaling network. For example, both loss- and gain-of-functions of MeCP2 in mice were shown to cause similar hippocampal circuit dysfunction [19]. Alternatively, one possible mechanism for this paradox is that LEOPARD syndrome associated loss-of-function PTPN11 mutations result in MAPK activation, not inactivation, by unknown mechanisms as mentioned above [1213].
PTPN11 mutations also induce cancers but, interestingly, mutations in cancer do not overlap with those in NS while mutations in both conditions commonly result in hyperactivation of the RAS-MAPK signaling pathway in most of the cases [20]. NS is a relatively common developmental disorder inherited in an autosomal dominant pattern and PTPN11 mutations account for 40%–50% of cases [5620]. The expressivity of NS is quite diverse and patients show a wide range of phenotypes including short stature, congenital heart deficits, typical craniofacial phenotypes, and various cognitive symptom [2021]. Not much attention has been paid to the cognitive features in NS compared to physical phenotypes. Recently, multiple studies have shown that cognitive problems, such as learning and memory impairments and social problems which substantially affect the quality of life, are relatively common among NS patients [222324]. Studies using animal models can provide in-depth understanding of the molecular and cellular mechanism underlying cognitive deficits in genetic disorders [252627]. There are multiple animal models of NS from fly to mouse [1218282930]. In this mini review, we will review the learning and memory phenotypes in NS mouse models and recent studies investigating the role of SHP2 in synaptic plasticity, with a specific emphasis on its regulation of glutamate receptors.
IMPAIRED LEARNING AND SYNAPTIC PLASTICITY IN Shp2 MUTANT MICE
In order to study the biological mechanism for the cognitive deficits in NS, several animal models have been generated. Pagani and colleagues [28] showed that flies expressing a constitutively active mutant Shp2 (csw) showed long-term memory deficits, which was one of the first reports demonstrating that Shp2 mutant animals display memory deficits. Mutant mice have been widely used as animal models of NS and other Rasopathies (for review, see [2529]). Shp2D61G/+ knock-in mice expressing a NS-associated gain-of-function mutant Shp2 showed phenotypes which are similar to typical symptoms of NS such as short stature and heart defects [18]. Shp2D61G/+ mice showed deficits in hippocampal learning and memory [3132]. Another knock-in mouse model expressing the most common NS allele Shp2N308D/+ also has impaired spatial learning and memory, albeit with a weaker phenotype than that seen in Shp2D61G/+ mice [31]. Memory deficits in Shp2D61G/+ mice can be reversed by reducing Erk activity in adult mice, suggesting that the increased activation of the Ras-Erk signaling pathway is responsible for the memory deficits in the mutant mice [31]. Interestingly, a recent study showed that overexpressing the mutant Shp2D61G only in hippocampal excitatory neurons, but not in inhibitory neurons, can induce spatial memory deficits in mice [33]. Synaptic plasticity is considered as a cellular mechanism of higher cognitive functions including learning and memory [34353637]. Consistently, both Shp2D61G/+ and Shp2N308D/+ models show significant deficits in long-term potentiation (LTP) in the hippocampal Schaffer-collateral (CA3-CA1) pathway, which can be also reversed by reducing Erk activation [31], strongly suggesting that LTP deficits may underlie the memory deficits in NS.
The role of Shp2 in memory and synaptic plasticity has also been examined in a conditional knockout mouse in which Shp2 is specifically deleted in the forebrain using αCaMKII-CRE [38]. The conditional knockout mice exhibited a mild deficit in spatial learning in the Morris water maze task [38]. Basal synaptic transmission and post-tetanic potentiation at CA3-CA1 synapse are significantly reduced, but hippocampal LTP is normal in the knockout mice [38], suggesting that Shp2 is required for normal synaptic transmission and short-term synaptic plasticity in the hippocampus. Interestingly, another conditional Shp2 knockout mouse line (αCaMKII-Cre: Shp2flox/flox or CaSKO), in which Shp2 is also deleted by αCaMKII-Cre, showed deficits in long-term fear memory tested 7 days after training, whereas recent memory tested 1 day after training was intact [39]. Furthermore, this knockout showed significant impairment in hippocampal LTP [39]. The discrepancy between these two knockout mice might stem from differences in either deleted exons (exon 4 or 11) or the CRE lines used in each study [4041]. Studies using Shp2 knockout and Shp2 knock-in mice reviewed here demonstrate that Shp2 is critically involved in learning and memory, as well as synaptic plasticity. But what is the mechanism underlying synaptic plasticity deficits in NS mouse models?
Shp2 AND THE NMDA RECEPTOR
Glutamate is a major neurotransmitter in the central nervous system and its receptors play critical roles in regulating synaptic plasticity such as LTP and long-term depression (LTD) [34353642]. Among glutamate receptors (GluRs), N-methyl-D-aspartate (NMDA) receptors function as a coincidence detector which is crucial for the induction of LTP and LTD [34]. Accordingly, blocking NMDA receptors (NMDARs) impairs both synaptic plasticity and learning [434445]. NMDARs interact with multiple proteins including protein kinases and phosphatases [46]. Shp2 has also been found to interact with GluN2B (Grin2b or NR2B) in mouse and rat brain [4647]. Nerve ligation induced the expression of GluN2B, phosphorylation of GluN2B (p-GluN2B) at Y1472, as well as the interaction between PSD-95 and phosphorylated GluN2B (p-GluN2B) in rat dorsal horn [48]. Interestingly, the upregulation of GluN2B and its interaction with PSD-95 are blocked by Shp2 inhibition, suggesting that Shp2 is involved in regulating GluN2B phosphorylation and its interaction with PSD-95 [48]. Later, it was shown that BDNF treatment increases the phosphorylation of Shp2, as well as the interaction between Shp2 and GluN2B, in cortical neurons and spinal cord of rats [4749]. By an as yet unknown mechanism, phosphorylated Shp2 enhances the functional expression of GluN2B and positively regulates LTP induction in rat spinal cord after spinal nerve ligation [49]. In addition, inhibiting Shp2 by siRNA or a pharmacological inhibitor blocks BDNF- or spinal nerve ligation-induced LTP, suggesting that Shp2 is required for GluN2B-dependent LTP in rat spinal cord [49].
Shp2 is also involved in regulating NMDAR function in the forebrain. Phosphorylation of GluN2A (Grin2a or NR2A) at Y1325 and GluN2B at Y1472, which are Src phosphorylation target sites, were increased in the hippocampus of Shp2 knockout (CaSKO) mice that showed reduced mEPSC frequency and impaired LTP in the hippocampus [39]. In the brain, Yan and colleagues showed that the synaptic expression or function of the zinc-sensitive GluN2A subunit of NMDAR was increased in pyramidal neurons of the hippocampal CA1 region in CaSKO mice, whereas the NMDAR/AMPAR current ratio in CaSKO mice was not different from that of wild type controls [39]. It remains unclear how the alteration in subunit composition of synaptic NMDAR leads to deficits in LTP in CaSKO mice, without affecting overall NMDAR currents in the hippocampus.
A recent study has shown that NMDAR function is altered in Shp2D61G/+ mice [32]. Levy and colleagues found that NMDAR-, but not AMPAR-mediated, current is significantly reduced in the hippocampal neurons of Shp2D61G/+ mice ([32]; but also see below). Specifically, Shp2 was found to directly dephosphorylate GluN2B Y1252, which interrupts the binding of GluN2B to the actin regulatory scaffolding protein Nck2 [32]. NMDAR-mediated current decayed faster in Shp2D61G/+mutants than in wild type littermates and NMDAR-current in Shp2D61G/+ mice were insensitive to the GluN2B blocker ifenprodil, suggesting that GluN2B function is reduced in Shp2D61G/+ mice [32]. These results might be inconsistent with the previous finding that Shp2 positively regulates NMDAR function in spinal cord [49]. In addition, surface expression of GluN2B was not reduced in mutant mice and it remains to be investigated how the direct dephosphorylation of GluN2B Y1252 by Shp2 reduces GluN2B function.
Shp2 AND THE AMPA RECEPTOR
In addition to NMDAR, α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptor (AMPAR) is a subtype of ionotropic glutamate receptors which also plays critical roles in synaptic plasticity [5051]. Regulations of the postsynaptic membrane trafficking of AMPAR are the primary mechanism for the expression of LTP and LTD [5253]. Kinases and phosphatases, in general, bi-directionally regulate the synaptic plasticity (LTP vs. LTD) by either directly or indirectly regulating AMPAR trafficking [545556]. Among phosphatases, protein phosphatase 1 (PP1) and calcineurin have been implicated in LTD induction [57]. Intriguingly, in contrast to the other phosphatases, recent studies strongly suggest that Shp2 is required for the synaptic delivery of AMPAR as well as LTP induction. LTP induction either by chemical or electrical stimulation increased Shp2 activity assessed by its Y542 phosphorylation, GluA1 (GluR1 or Gria1) phosphorylation at S845 and GluA1 surface expression in the hippocampal neurons [58]. A Shp2 inhibitor NSC87877 suppressed the phosphorylation of Shp2 as well as the phosphorylation and membrane trafficking of GluA1, suggesting that Shp2 positively regulates AMPAR trafficking during LTP induction [58]. Notably, MEK inhibitor U0126 treatment blocked LTP-induced phosphorylation of GluA1 S845 without affecting Shp2 phosphorylation, suggesting that ERK is a downstream of Shp2 [58]. More recently, it has been shown that Shp2 phosphatase activity is critically involved in regulating the phosphorylation of GluA1 at S845 and its surface expression during the TTX-mediated synaptic upscaling [59]. Since the prolonged TTX treatment was shown to decrease ERK1/2 activity [59], it is unlikely that Shp2 regulate GluA1 phosphorylation via Ras-Erk pathway. How Shp2 regulates GluA1 phosphorylation remains to be further investigated.
A NS associated mutant Shp2 (Shp2D61G) was also shown to increase AMPAR trafficking. Overexpressing Shp2D61G in fully matured cultured hippocampal neurons (DIV 21) significantly increased the number of the surface GluA1 clusters, which was blocked by inhibiting MAPK activity [3160]. Consistently, the frequency of excitatory postsynaptic current (EPSC) and AMPA/NMDA current ratio were significantly higher in the hippocampal pyramidal neurons of both Shp2D61G/+ knock-in mice and AAV-Shp2D61G expressing mice compared to the control groups, which were reversed by reducing MAPK activity [3133]. Taken together, these results strongly suggest that Shp2 can facilitate the synaptic trafficking of GluA1 in the hippocampus. However, the mechanism of how Shp2 regulates the surface expression of GluA1 remains to be investigated.
PERSPECTIVES
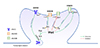
We reviewed that Shp2 is involved in regulating glutamate receptors. However, it is still not clear how Shp2 regulates glutamate receptors. Shp2 may have both direct and indirect role in regulating the function and expression of glutamate receptors (Fig. 1). Considering that Shp2 is generally acting as a positive regulator of the RAS-ERK pathway, Shp2 may indirectly facilitate AMPAR synaptic trafficking through activating the RAS-ERK pathway. The activation of the RAS-ERK pathway has been shown to facilitate AMPAR trafficking to the synaptic membrane [61]. For example, expressing a constitutively active RAS mutant enhanced the surface expression of AMPAR [62]. Inhibiting Shp2 by NSC87877 treatment blocked both ERK phosphorylation and AMPAR trafficking during chemical LTP induction in hippocampal cultures [58]. The Shp2D61G mutant enhanced AMPAR-mediated current in the hippocampus, which was fully reversed by MEK inhibitor SL327 treatment [31], suggesting that there is a strong correlation between Shp2, ERK, and AMPAR trafficking.
In addition to the suggested role of Shp2 in facilitating AMPAR trafficking via ERK activation, Levy and colleagues showed that Shp2 directly dephosphorylates GluN2B at Y1252 without affecting the phosphorylation at Y1472 or Y1336, in both in vitro and ex vivo conditions [32]. This direct dephosphorylation of GluN2B at Y1472 reduces the localization of the scaffolding protein Nck2 to dendritic spines and contributes to the reduction of GluN1-GluN2B mediated currents without affecting AMPAR-mediated currents [32]. However, it is still controversial whether Shp2 directly regulates NMDAR function in the hippocampus. As reviewed above, AMPAR, but not NMDAR expression or function, was increased in the adult hippocampus or mature hippocampal cultures expressing Shp2D61G [3160]. Interestingly, the mutant Shp2D61G was shown to differentially regulate AMPAR and NMDAR surface expression depending on developmental maturation [60]. Expressing Shp2D61G in premature hippocampal neurons (DIV 6) only increased the surface expression of the GluN1 subunit of NMDAR without affecting AMPAR, whereas expressing Shp2D61G in mature neurons (> DIV 12) increased GluA1 expression without affecting GluN1 expression [60]. These age-dependent distinct roles of Shp2 in glutamate receptor trafficking may underlie the seemingly contradictory findings (increased vs. decreased AMPA/NMDA currents in Shp2D61G/+ mice) from different laboratories [3132]. In addition, the difference in genetic backgrounds of the mice (mixed C57BL/6J and 129/SvJ background vs. 129S6/SvEv) might also have contributed to the distinct phenotypes [3132].
There are also inconsistent results on the role of Shp2 on GluN2B Y1472. Peng and colleagues [48] showed Shp2 activity was required for the phosphorylation of GluN2B at Y1472 in rat dorsal horn after nerve ligation by an unknown mechanism. However, in the forebrain, GluN2B Y1472 phosphorylation was significantly increased in Shp2 knockout mice [39]. The increased GluN2B Y1472 phosphorylation was reversed by a Src family kinase inhibitor, suggesting that Shp2 may negatively regulate GluN2B Y1472 phosphorylation through suppressing Src activity [39]. This result is interesting because previous studies have shown that Shp2 is involved in activating Src family kinases including Src [56364]. Differences in tissue or cell type (dorsal horn vs. forebrain or fibroblast vs. neurons) may be responsible for these contradictory results. Indeed, it has been shown that the impacts of Shp2 mutation have cell type and context-specificity [1830333964]. Recently, Ryu and colleagues showed that the SHP2D61G mutant increases Ras-Erk activation only in excitatory, but not in inhibitory, neurons in adult mouse hippocampus [33]. Therefore, it is plausible to argue that Shp2 may recruit distinct downstream signaling pathways to regulate the function and expression of glutamate receptors in a cell type- and age-specific manner. Understanding the cell type-specific role of SHP2 is critical when developing treatments for SHP2-associated disorders such as NS and LEOPARD syndrome, since SHP2 is ubiquitously expressed in the brain [386566]. Advances in single cell biology such as single cell genomics combined with either whole cell patch clamp recording or state-of-the-art imaging techniques using molecular sensors for Shp2, Erk, or glutamate receptors will be useful in answering these remaining questions.
 XML Download
XML Download