

 PDF
PDF Citation
Citation Print
Print



Coronaviruses (CoVs), within the order Nidovirales, are enveloped, single-strand, positive-sense RNA viruses with a large genome of approximately 30 kbp in length. CoV was cultured for the first time in human embryonic tracheal organ cultures by Tyrrell and Bynoe in 1965,1 and it was named as ‘corona’ due to crown-like appearance of the surface projections on electron microscopy. All CoVs develop only in the cytoplasm of infected cells, bud into cytoplasmic vesicles, and then extrude in virus particles of 70–80 nm in diameter by exocytic secretory pathway.
Among four genera of CoVs, beta-CoV includes five subgenus—embevovirus, sarbecovirus including severe acute respiratory syndrome (SARS)-CoV, merbecovirus including Middle East respiratory syndrome (MERS)-CoV, nobecovirus, and hibecovirus. Because CoVs can infect a variety of animals, SARS-CoV and MERS-CoV crossed the species barriers.
Since December 2019, 2019 novel CoV (SARS-CoV-2) has been making a large outbreak involving 49,053 laboratory-confirmed patients and 1,383 mortality in 25 countries including Korea until February 14, 2020.2 In the outbreak situation, isolation of causative virus is indispensable for developing and evaluating diagnostic tools, therapeutics, and vaccine candidates. SARS-CoV-2 was first isolated using human airway epithelial cells and it was classified into the subgenus sarbecovirus of beta-CoVs by phylogenetic analyses of the gene sequences.3 Both the SARS-CoV and the MERS-CoV were initially isolated and grew readily in Vero cells.45 Here, we report the isolation of SARS-CoV-2 using Vero cells from a patient entering Korea from Wuhan, China.
The patient with the first laboratory-confirmed SARS-CoV-2 infection in Korea is published previously.6 Briefly, a 35-year-old woman developed fever, chill, and myalgia on January 18, 2020, and arrived at the Incheon airport from Wuhan on the next day. After laboratory-confirmed diagnosis of SARS-CoV-2 infection, she developed nasal congestion, cough, and sputum. Oxygen supplementation was started on day 4 of her illness, and her oxygen requirement increased to 6 L/min on day 7 of illness. Fever persisted for ten days and her maximum body temperature during her illness was 38.9°C on day 7 of illness.
The patient's oropharyngeal samples were obtained by using UTM™ kit containing 1 mL of viral transport media (Copan Diagnostics Inc., Murrieta, CA, USA) on day 7 of her illness. We inoculated monolayers of Vero cells (ATCC
® CCL-81™) with the samples and cultured the cells at 37°C in a 5% carbon dioxide atmosphere. Until 5 days after inoculation, cytopathic effects were not distinct, which is compatible with the previous findings that no specific cytopathic effects were observed in the Vero E6 cells until 6 days after inoculation in the report about first isolation of SARS-CoV-2.3 Five days after inoculation, we did blind passage of culture supernatant into T-25 culture flask (ThermoFisher Scientific Inc., Waltham, MA, USA) with monolayers of Vero cells, and cytopathic effects consisting of rounding and detachment of cells were observed in the whole area of the T-25 flask 3 days after the first blind passage (Fig. 1A and B).
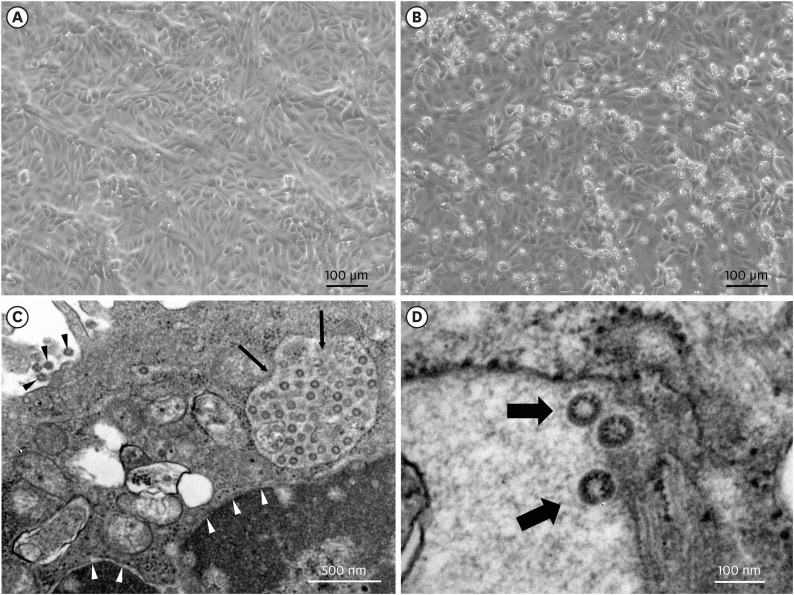 | Fig. 1Cytopathic effects of SARS-CoV-2 in Vero cell cultures and electron microscopy image of SARS-CoV-2. Vero cells were inoculated with oropharyngeal swab sample. (A) Vero cell cultures in negative control. (B) Cytopathic effects consisting of rounding and detachment of cells in Vero cell cultures 3 days after the first blind passage. (C, D) Transmission electron microscopy image of Vero cells infected with SARS-CoV-2. White arrow head denotes nuclear membrane, black arrow head extracellular virus particles, and thin black arrow cytoplasmic vesicle including virus components (C). Thick black arrow denotes magnified virus particles with crown-like spikes (D).SARS = severe acute respiratory syndrome, CoV = coronavirus.
|
In order to observe virus particles, Vero cell monolayer showing the cytopathic effects was fixed as previously described.7 It was cut on ultramicrotome (RMC MT-XL; RMC Boeckeler, Tucson, AZ, USA) at 65 nm. Ultrathin sections were stained with saturated 4% uranyl acetate and 1% lead citrate before examination with a transmission electron microscope (JEM-1400; JEOL USA Inc., Peabody, MA, USA) at 80 kV. Spherical particles with crown-like spikes ranging 66 to 81 nm in diameter were observed within the cytoplasmic vesicles and in the extracellular space adjacent to cell membrane (Fig. 1C and D).
For whole genome sequencing of the virus isolate (BetaCoV/Korea/SNU01/2020), culture supernatant of Vero cells infected was used for RNA extraction. RNA was extracted by using QIAamp viral RNA mini kit (QIAGEN, Valencia, CA, USA), according to the manufacturer's instructions. RNA libraries were prepared using TruSeq Stranded Total RNA Kit (catalog No. 20020596; Illumina, San Diego, CA, USA) according to the manufacturer protocol. Sequencing was performed on an Illumina Nextseq 500 platform, produced on average a total of 150 million reads, 150 bp per sample, as per the manufacturer's instructions in Macrogen Inc. (Seoul, Korea).8910 FASTQ was used to trim the adapter and remove low quality bases and reads. Qualified reads were mapped to NC_045512, a SARS-CoV-2 genome reference using Burrows-Wheeler Aligner (v0.7.12-r1039), and a bam file was produced.11 In this bam file, the variation was confirmed by comparing with genome using SAMtools (v1.3.1).12 For genome-base phylogeny analysis, 37 strains including BetaCoV/Korea/KCDC03/2020 were used in combination with BetaCoV/Korea/SNU01/2020. The sequences used for analysis were downloaded from NCBI (http://www.ncbi.nlm.nih.gov/) and GISAID (http://www.gisaid.org). The 37 strain genomes were multiple-sequence aligned using MAFFT (v7.450), a sequence alignment tool, and were used to generate phylogenetic tree.13 Phylogenetic analysis of the aligned sequence was performed with 1,000 bootstrap replicates using MEGAX and a general time-reversible model used as the nucleotide substitution model.14
Next-generation sequencing of BetaCoV/Korea/SNU01/2020 (GenBank accession no. MT039890) revealed 9 mutations compared to the NC_045512 reference genome isolated from Wuhan (Table 1). Most of the mutations in our isolate consisted of 70% alternative genes and 30% reference genes (NC_045512). Five variants were found in ORF1ab, one variant in S gene, two variants in ORF3a, and one variant in E gene. Of the nine mutations, six also showed changes in amino acids. When comparing our isolate with the one isolated from the Korea Centers for Disease Control and Prevention (BetaCoV/Korea/KCDC03/2020), 12 variants including the above 9 mutations were found. These mutations may occur by cell culture-adaptation in that our culture isolate was obtained after first blind passage, or by micro-evolution of SARS-CoV-2 before acquisition in Wuhan. Because those genome sequences are quite homologous each other, it is difficult to validate these two hypothesis.
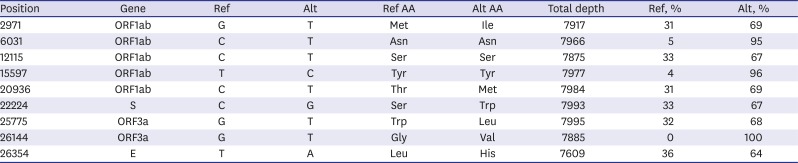
Table 1
Genetic variations of our isolate (BetaCoV/Korea/SNU01/2020) compared to the NC_045512 reference genome of a SARS-CoV-2 isolated in Wuhan
SARS = severe acute respiratory syndrome, CoV = coronavirus, Ref = reference, Alt = alternative, AA = amino acid.
![]()
The phylogenetic tree was analyzed using 37 genome information including CoVs isolated from Korea and Wuhan (Fig. 2). As a result, it was confirmed that our isolate is a beta-CoV belonging to the subgenus sarbecovirus, and that it closely clustered with other SARS-CoV-2 isolated from Korea and Wuhan.
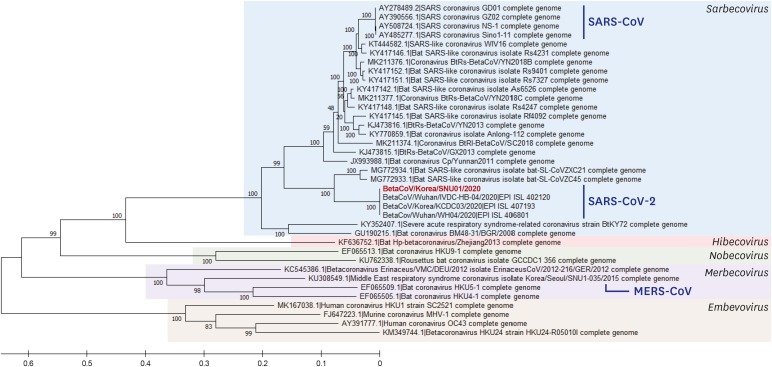
In summary, we isolated SARS-CoV-2 using Vero cells from the first laboratory-confirmed SARS-CoV-2-infected patient in Korea. Phylogenetic analyses of the whole genome sequences showed that it clustered with other SARS-CoV-2 reported from Wuhan, China.
 XML Download
XML Download