

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
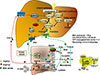
The global epidemic of obesity has caused the increase of the prevalence of type 2 diabetes mellitus (T2DM), which is predicted to increase 54% by 2030 [1]. T2DM is a complex metabolic disease developed most often in middle-aged and older people with a family history of diabetes and obesity. Asian, Hispanic, American Indian, and African American populations have high prevalence of T2DM. The majority of diabetes patients (70%) are overweight or obese, and the obesity rate is expected to increase 33% in the next 20 years [2]. Diabetic and obese patients have increased risk for cardiovascular disease (CVD), the leading cause of death in western countries (Fig. 1). T2DM patients are insulin resistant and glucose intolerant in skeletal muscle, adipose tissue, and liver. Consequently, hyperglycemia induces vascular complications in blood vessels, kidney, heart, and liver, and can cause renal and retinal disorders and macrovascular complications, leading to blindness, kidney failure, atherosclerosis, stroke, and liver diseases.
T2DM is linked to non-alcoholic fatty liver disease (NAFLD) [3], which has rapidly increased worldwide and has a global prevalence of about 24%; the highest rates being in South America, the Middle East, Asia, the USA, and Europe [4]. NAFLD is a significant complication of obesity and diabetes [5] and is an independent risk factor for CVD (Fig. 1) [678]. CVD and NAFLD are the heart and liver manifestations of the metabolic syndrome (syndrome X) [9], a collection of five abnormal metabolic phenotypes: hypertension, hyperglycemia, hypertriglyceridemia, insulin resistance, and obesity (Fig. 1) [10]. NAFLD consists of a broad spectrum of liver diseases from simple hepatic steatosis and nonalcoholic steatohepatitis (NASH) to cirrhosis and hepatocellular carcinoma (HCC), the end stages of liver disease [111213]. High fat, high carbohydrate, or high calorie diets, alcohol, starvation, drugs, and viral infection can cause hepatic steatosis [14]. Simple steatosis is reversible, about 30% of NAFLD patients progress to NASH, a chronic condition with liver inflammation, macrovascular ballooning, macrophage infiltration and fibrosis. About 2% to 5% of NASH patients develop liver cirrhosis and HCC. NASH has become the second leading cause of liver cancer and will surpass viral hepatitis as the primary cause for liver transplants. Multiple factors, including reactive oxidizing species, visceral fat, viruses, cigarette smoking, insulin resistance, lipotoxicity, and hepatic cholesterol have been linked to the progression of simple hepatic steatosis to NASH [15], and there is no U.S. Food and Drug Administration-approved therapeutic drug for treating NASH.
Bile acids are derived from cholesterol catabolism in the liver and are amphipathic molecules with strong detergent properties that aid in the absorption of dietary fats and steroids, lipid-soluble vitamins, and xenobiotics, including drugs and environmental contaminates. Bile acids are now recognized as key endogenous steroid molecules that play critical roles in regulating and maintaining lipid, glucose and energy metabolism, protecting against inflammation in the liver, intestine and heart, and preventing diabetes and obesity [16]. Bile acid metabolism is altered in T2DM [171819] and dysregulation of lipid, glucose and energy metabolism causes inflammatory metabolic diseases including T2DM, NAFLD, and CVD. This review will cover bile acid pathophysiology and signaling, recent advances in understanding the role of bile acid signaling in diabetes, and bile acids as drug therapies for treating metabolic diseases.
BILE ACID BIOLOGY AND PHYSIOLOGY
Bile acid biology
Bile acids are the end products of cholesterol catabolism in the liver, the only organ which has all the enzymes required in the cascade pathway to convert excessive cholesterol to bile acids. Bile acid synthesis accounts for catabolism of about 50% of the daily cholesterol output. Biliary secretion of cholesterol in bile accounts for another 40% of the daily cholesterol output. The remaining 10% of cholesterol is utilized for membrane synthesis and steroid hormone synthesis in steroidogenic tissues. Therefore, bile acid metabolism plays a critical role in maintaining whole body cholesterol homeostasis.
Bile acid synthesis in the liver
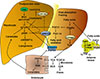
In the human liver, cholesterol 7α-hydroxylase (CYP7A1) catalyzes the first and rate-limiting step in the classic bile acid synthesis pathway and synthesizes two primary bile acids, chenodeoxycholic acid (CDCA) and cholic acid (CA), the latter of which requires sterol 12α-hydroxylase (CYP8B1) (Fig. 2). CA has 3 hydroxyl (HO) groups at the 3α, 7α, and 12α-positions; thus, it is more soluble (hydrophilic) than CDCA, which has 2 hydroxyl groups at the 3α and 7α-positions. All hydroxyl groups in bile acids are facing one side of the carbon skeleton, creating a hydrophilic side and a hydrophobic side, making bile acids amphipathic molecules with strong detergent properties. Mitochondrial sterol 27-hydroxylase (CYP27A1) then catalyzes steroid side-chain oxidation, followed by a peroxisomal β-oxidation reaction that cleaves a 3-carbon unit from the steroid-side chain to form C-24 bile acids and propionyl-CoA. Bile acids also can be synthesized via the alternative pathway, initiated by CYP27A1, to form 27-hydroxycholesterol and 3β-hydroxy-5-cholestenoic acid, which is then hydroxylated at the 7α-position by the non-specific oxysterol 7α-hydroxylase (CYP7B1). CYP27A1 and CYP7B1 are expressed in most tissues and macrophages and are responsible for the metabolism of oxysterols to steroid hormones in steroidogenic tissues; these oxidized steroid intermediates can be transported to the liver for synthesis of bile acids. The classic pathway is the major route for bile acid synthesis in humans. In rodents, most CDCA (3α, 7α) is converted to α-muricholic acid (α-MCA, 3α, 6β, 7α) and the 7α-HO group is epimerized to 7β-HO by Cyp2c70 (sterol 6β-hydroxylase), forming β-muricholic acid (β-MCA, 3α, 6β, 7β). Addition of a 6β-HO group to CDCA converts hydrophobic CDCA to highly soluble and non-toxic α-MCA and β-MCA. In contrast to humans, CA (50%) and α-MCA plus β-MCAs (50%) are the predominant primary bile acids produced in mouse liver [20].
Most bile acids are conjugated to glycine (G) and taurine (T) in a ratio of about 3:1 in humans. In mice, most bile acids (>95%) are taurine-conjugated. The conjugated bile acids are secreted into bile and stored in the gallbladder, and after meal intake, bile acids are secreted into the intestinal tract. Bile acids are reabsorbed, mostly in the terminal ileum and colon, and are secreted into portal blood circulation back to the liver to inhibit bile acid synthesis. This enterohepatic circulation of bile acids from the liver to intestine and back to the liver occurs six to eight times a day and is highly efficient in reabsorbing about 95% of bile acids in a pool of about 10 g in an average human. Small amounts of bile acids lost in feces (5%, 0.5 g/day) are replenished by de novo synthesis in the liver (Fig. 2) [20].
Bile acid biotransformation in the gut
The gut bacteria metabolize primary bile acids to secondary bile acids, which were once considered “damaged” bile acids that were excreted into feces or cleared in urine. In the intestine, a portion of conjugated CA and CDCA are de-conjugated by gut bacterial bile salt hydroxylase (BSH) to free bile acids, then bacterial 7α-dehydroxylase activity removes a 7-HO group from CA and CDCA to form deoxycholic acid (DCA) and lithocholic acid (LCA), respectively (Fig. 2) [21]. LCA is a toxic and highly insoluble bile acid, most of which is excreted into feces, though small amounts of LCA (approximately 2%) are circulated to the liver and sulfoconjugated for secretion into urine. DCA is a potent bactericide that controls bacterial overgrowth, but also is a promoter of colon cancer. Small amounts of CDCA (1% to 2%) are converted to its 7β-epimer, ursodeoxycholic acid (UDCA) by gut bacterial 7β-hydroxysteroid dehydrogenase in humans. Epimerization of the C7-HO group from the α- to the β-position converts toxic CDCA to hydrophilic and non-toxic UDCA. In humans, the circulating bile acid pool is highly hydrophobic, consisting of CA, CDCA, and DCA in a ratio of about 40:40:20, and the ratio of glycine to taurine-conjugated bile acids is about 3 to 1 [20].
BILE ACID SIGNALING IN METABOLIC REGULATION
Extensive research in the last three decades has identified bile acids as signaling molecules that activate several nuclear receptors: farnesoid X receptor (FXR) [222324], vitamin D receptor (VDR) [25], pregnane X receptor (PXR) [26]; and the membrane G protein-coupled receptors: Takeda G protein-coupled receptor 5 (TGR5) [27], sphingosine-1 phosphate receptor 2 (S1PR2) [28], and muscarinic M2 receptor [29]. These bile acid-activated receptors play critical roles in liver metabolism [30]. This section will focus on the roles of FXR and TGR5 in the regulation of metabolism and pathophysiology of liver-related metabolic diseases.
Farnesoid X receptor
FXR is mainly expressed in the digestive system, including liver and intestine. FXR is activated by bile acids in the order of potency CDCA>LCA=DCA>CA. FXR knockout mice have increased hepatic triglycerides, cholesterol and a proatherogenic lipid profile, and reduced bile acid pool and increased fecal bile acid secretion, indicating FXR plays a major role in bile acid and lipid metabolism [31]. FXR also regulates the enterohepatic circulation of bile acids and feedback homeostasis [32]. In the liver, bile acids activate FXR to induce the expression of the major hepatic bile acid efflux transporter, bile salt export pump which secretes conjugated bile acids into bile, and inhibits the sinusoidal hepatic bile acid uptake transporter, Na+2-dependent taurocholate co-transport peptide (Fig. 2). These two major bile acid transporters regulate hepatic bile acid homeostasis. In hepatocytes, bile acid activation of FXR induces a transcriptional repressor, small heterodimer partner, to inhibit transcription of the CYP7A1 and CYP8B1 genes (Fig. 2). In the ileum, bile acids are reabsorbed into enterocytes via apical sodium-dependent bile acid transporter, whose function is inhibited by bile acids. Bile acids activate intestinal FXR to induce the release of the intestinal hormone fibroblast growth factor 19 (FGF19) in humans or FGF15 in mice (Fig. 2). FXR also induces the bile acid efflux transporters organic solute transporter α and β (OSTα/OSTβ) to secrete bile acids into portal blood circulation. FGF19 released from enterocytes is transported via portal blood circulation to hepatocytes and binds to the membrane FGF receptor 4/β-Klotho complex, which activates mitogen-activated protein kinase (MAPK) and extracellular regulated kinase 1 and 2 (ERK1/2) signaling to inhibit CYP7A1 and CYP8B1 gene transcription (Fig. 2). The intestinal FXR/FGF19 to hepatic FGFR4 pathway may be the major physiological mechanism for bile acid feedback regulation of bile acid synthesis.
The role of FXR in glucose metabolism is controversial. It was reported that FXR expression was reduced in streptozotocin-induced diabetic rats, and insulin and glucose induced FXR expression [33]. Activation of FXR has been shown to improve glucose and lipid metabolism and reduce inflammation in diabetes [343536]. Surprisingly, other studies reported that Fxr−/− mice had improved hyperglycemia and insulin sensitivity, and activation of FXR induced obesity and diabetes by reducing energy expenditure [37]. In pancreatic β-cells, activation of FXR stimulates glycolysis to increase the adenosine triphosphate:adenosine diphosphate (ATP:ADP) ratio and results in closing KATP-channels and depolarizing the plasma membrane, which subsequently opens Ca2+ channels, increasing Ca2+ influx and stimulating insulin secretion from β-cells [38].
Takeda G protein-coupled receptor 5
In the liver, TGR5 is expressed in sinusoidal endothelial cells, Kupffer cells (hepatic resident macrophages), stellate cells, and biliary epithelial cells in bile ducts, but not in hepatocytes [394041], and the secondary bile acids LCA and DCA are potent endogenous TGR5 agonists (LCA>DCA>CDCA>CA). TGR5 is expressed in the epithelium of human gallbladder and controls gallbladder refiling [42]. TGR5 also plays a key role in bile acid metabolism and fasting-induced hepatic steatosis [43]. In the colon, TGR5 mediates bile acid-induced gastrointestinal motility, transit time and defecation [44]. In the intestine and macrophage, activation of TGR5 protects against inflammation [45]. TGR5 also plays a critical role in the control of glucose homeostasis [46]. Activation of TGR5 stimulates the release of glucagon-like peptide-1 (GLP-1) from enteroendocrine L-cells to stimulate insulin secretion from β-cells and increase insulin sensitivity. In adipose tissue, activation of TGR5 induces thyroid hormone deiodinase type 2 (DIO2), which converts thyroid hormone thyroxine (T4) to triiodothyronine (T3) to stimulate energy metabolism and white adipose tissue browning (Fig. 2). CDCA increases brown adipose tissue in humans, likely through TGR5-mediated increase of uncoupling protein and DIO2 expression [47]. TGR5 knockout mice are protected from cholesterol gallstone disease and high fat diet (HFD) induced obesity [4849]. FXR and TGR5 are co-expressed in L-cells, and activation of intestinal FXR stimulates TGR5 gene transcription via an FXR response element located in the TGR5 gene promoter; this crosstalk stimulates GLP-1 secretion [50]. It is therefore likely that some of the reported FXR effects on glucose metabolism may be due to TGR5 signaling in the gut.
BILE ACID SIGNALING IN DIABETES
Bile acids are nutrient sensors and metabolic regulators
Bile acid synthesis and CYP7A1 expression exhibit circadian rhythms, which are modulated by fasting and feeding. Sleep disruption, alcohol, and HFD disrupt these rhythms and cause altered bile acid homeostasis, contributing to the pathogenesis of insulin resistance and obesity [2051525354]. Bile acids are metabolic sensors that aid in dietary nutrient absorption to ultimately provide fuel for energy metabolism and biosynthesis.
Post-prandial regulation
Feeding rapidly stimulates the release of bile acids stored in the gallbladder and de-represses CYP7A1 expression to stimulate bile acid synthesis during the postprandial state. Feeding inhibits CYP8B1 expression, reducing CA and increasing CDCA, which is a more potent FXR agonist than CA. In the postprandial state, bile acids activate FXR to inhibit hepatic lipogenesis by stimulating insulin and insulin receptor substrate 1 (IRS1)-AKT-phosphoinositide 3-kinase (PI3K) signaling, which inhibits mechanistic target of rapamycin complex 1 (mTORC1) and induces autophagy (Fig. 3) [5556]. mTORC1-pS6K promotes maturation and nuclear localization of steroid regulatory element binding protein 1c (SREBP1c) to stimulate lipogenesis [57]. FXR inhibition of the mTORC1-SREBP1c pathway may be the most plausible mechanism for bile acid inhibition of lipogenesis.
Fasting and starvation
During fasting, serum insulin and glucose levels decrease while glucagon increases to induce gluconeogenesis and release glucose from glycogen to prevent hypoglycemia. Glucagon stimulates hormone sensitive lipase A to release free fatty acids from triglycerides in adipose tissue (Fig. 3). During prolonged fasting and starvation, free fatty acids activate peroxisome proliferator-activated receptor α (PPARα) to induce FGF21 and peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α) in liver and adipose tissue [6061]. FGF21 is a nutrient sensor that stimulates insulin sensitivity and regulates glucose and energy metabolism in adipose tissue. It also reduces serum triglycerides in diet-induced obese mice [60626364] and inhibits mTORC1 signaling in hepatocytes (Fig. 3) [63]. Fasting induces CYP8B1, resulting in increased CA and DCA in the bile acid pool, which induces ceramide synthesis via FXR and activates hepatic mTORC1 signaling to stimulate lipogenesis and cause hepatic insulin resistance (Fig. 3) [65].
Hyperglycemia
In hyperglycemia, glucose is converted to acetyl-CoA, which stimulates histone acetylation. Glucose and insulin are known to stimulate CYP7A1 gene transcription by increasing histone acetylation of CYP7A1 chromatin [6667]. This effect is referred to “glucose memory”. In diabetic and obese patients, stimulation of bile acid synthesis increases serum bile acids, with a higher ratio of 12α-hydroxylated bile acids (CA and DCA) to non-12α-hydroxylated bile acids (CDCA) [1819]. Increased CYP8B1 and CA stimulates dietary fat and cholesterol absorption and may contribute to dyslipidemia, diabetes, and obesity. Inhibition of CA synthesis improves glucose homeostasis and prevents diet-induced obesity and atherosclerosis in mouse models [686970].
INTERACTIONS BETWEEN BILE ACIDS AND THE GUT MICROBIOTA
Bile acids in the gut-to-liver axis
The gut-to-liver axis plays a critical role in the regulation of hepatic metabolism via the interactions between bile acids and the gut microbiota [717273]. Bile acid biotransformation by gut bacteria determines bile acid composition in the circulating bile acid pool and total bile acid pool size. Bile acids control gut bacterial growth, gut microbial composition, gut barrier function, and production of bacterial metabolites. Interactions between bile acids and gut bacteria significantly impact the health of the host and contribute to the pathogenesis of metabolic diseases, i.e., liver disease, obesity, and diabetes [74]. Dietary factors affect gut microbial growth and metabolism depending on protein, saturated or unsaturated fat, fiber, cholesterol, sucrose, fructose, and carbohydrate content [75]. Diet shapes the gut microbiota to alter host energy metabolism and contributes to diabetes and obesity. HFDs can cause gut dysbiosis and impair intestinal barrier function (leaky gut) by altering the gut microbiome. Gut microbes utilize short-chain fatty acids, mostly acetate, butyrate and propionate, for energy metabolism. Butyrate improves insulin sensitivity and energy metabolism [76] and Chinese T2DM patients had decreased butyrate-producing bacteria compared to healthy control subjects [77].
The gut microbiota in diabetes
The human gut microbiome consists of 3 trillion microorganisms in four major phyla, Firmicutes (60%), Bacteroidetes (22%), Actinobacteria (17%), and Proteobacteria (1%) [74]. A high ratio of Firmicutes to Bacteroidetes enables the gut microbiota to extract energy more efficiently from HFDs, increasing adiposity and obesity in humans. Animal-based diets increase the abundance of the bile-tolerant bacteria Bilophila wadsworthia and decreases Firmicutes [78]. High saturated fat diets or low fat diets supplemented with taurocholic acid (TCA) increase B. wadsworthia to promote a proinflammatory response and inflammatory bowel disease [79]. Increased abundance of Lactobacillus is associated with T2DM in humans [77] and in type 1 diabetes mellitus (T1DM) patients, Bacteroidetes increases and Firmicutes decreases. Higher Bacteroides species abundance is associated with autoimmunity, with increase of B. ovatus and B. uniformis, and decrease of B. fragilis in T1DM patients [75]. Interestingly, mucin-degrading Akkermansia muciniphila improves glucose tolerance in T1DM and HFD-fed mice [75].
Intestinal FXR in diabetes
T-α-MCA and T-β-MCA have been identified as antagonists of intestinal FXR that reduce bile acid feedback regulation and increase bile acid synthesis [71]. In germ-free mice, bile acid pool size is increased, with increased T-α-MCA and T-β-MCA and reduced TCA. Cyp8b1 deficiency also increased the bile acid pool in mice, with higher T-α-MCA and T-β-MCA, which antagonize intestinal FXR. These mice also had decreased hepatic lipogenesis, improved insulin tolerance, and altered gut microbiota [8081]. Intestinal FXR plays a critical role in metabolic disease via modulation of the microbiome [82]. Antibiotics, the antioxidant tempol, the FXR antagonist Gly-MCA, and deficiency of intestinal FXR all increase conjugated bile acids and T-β-MCAs, and suggest that inactivating intestinal FXR signaling decreases hepatic triglycerides in HFD-fed mice [8384]. Tempol decreases Lactobacillus, which has high BSH activity, and increases T-β-MCA content, which antagonizes intestinal FXR signaling and increases bile acid synthesis. Antagonizing intestinal FXR signaling also decreases circulating ceramides and inhibits de novo lipogenesis and hepatic gluconeogenesis [8485]. Fecal transplant of cecum microbiota from HFD-fed Fxr−/− mice and wild type mice into germ-free mice caused obesity [86]. These experiments indicate that FXR signaling may contribute to increased adiposity by altering the gut microbiota. A recent study reported that the anti-diabetic effect of metformin involved the reduction of B. fragilis and BSH activity [87]. This results in increased taurine (T)-UDCA and glycine (G)-UDCA, which antagonize intestinal FXR and improve hyperglycemia in diabetic patients. On the other hand, the intestine-specific FXR agonist fexaramine promotes adipose tissue browning and insulin sensitivity in mice [8889]. Fexaramine increases the LCA-producing gut bacteria Acetatifactor and Bacteroides, with both 7α- and 7β-dehydroxylase activities, to convert CDCA and UDCA to LCA. LCA stimulates TGR5/GLP-1 signaling to improve hepatic metabolism, and adipose tissue browning and energy metabolism [89]. Another study reported that gut commensal Bacteroides acidifaciens improved obesity and insulin sensitivity by increasing GLP-1 and decreasing intestinal dipeptidyl peptidase-4 (DDP-4) in mice [90]. This effect is apparently mediated by TGR5. Pro-hormone convertase 1/3 (PC1/3) cleaves preproglucagon to GLP-1, which is degraded by DDP-4. GLP-1 receptor agonists have been used to treat T2DM [91] and DDP-4 inhibitors (gliptins) reduce glucagon and increase serum GLP-1 levels and insulin sensitivity [92]. In contrast, another study indicated that activation of FXR in both enteroendocrine secretin tumor cells (STC-1) and in mice decreased proglucagon gene expression and GLP-1 secretion by interfering with carbohydrate responsive element binding protein gene expression and inhibiting glycolysis [93]. Therefore, intestinal FXR signaling can either aggravate or alleviate diabetes and obesity. It appears that FXR agonists and antagonists reshape the gut microbiota to exert differential effects on diabetes and obesity.
METABOLIC AND BARIATRIC SURGERY IMPROVES OBESITY AND DIABETES
Metabolic and bariatric surgery (MBG), such as Roux-en-Y gastric bypass (RYGB) and vertical sleeve gastrectomy (VSG), is the most effective way to reduce weight in overly obese patients [94]. Many recent studies report rapidly improved insulin sensitivity and diabetes 1 or 2 weeks following MBG, prior to weight loss, suggesting that metabolic changes are involved in improving glycemic control after gastric bypass. Serum total and primary bile acids, GLP-1, and FGF19 levels increase after MBG [9596979899], though the underlying mechanism for diabetes remission after MBG is not clear. Increased fasting serum bile acids, especially conjugated bile acids, implies a role for the gut microbiota in improving glycemic control and serum lipid profile in patients after gastric bypass [98].
The metabolic benefits of MBG may be mediated by FXR/FGF19 signaling and TGR5/GLP-1 signaling [100]. The increased serum bile acids and FGF19 in T2DM patients after RYGB may indicate a dysregulation of the CYP7A1-FGF19 negative feedback pathway [101]. In obese human patients, circulating FGF21 is paradoxically induced, indicating FGF21 resistance, and bariatric surgery reduces serum FGF21 [102].
In Fxr−/− and Tgr5−/− mice, the effect of VSG on glucose tolerance was reduced [103104]. Biliary diversion to the ileum also resulted in metabolic effects similar to RYGB in Tgr5−/−, but not intestine-specific Fxr−/− mice, suggesting a role of intestinal FXR in bile acid- and GLP-1-mediated metabolic improvement following bariatric surgery [105]. This study also suggested that intestinal bile acids and A. muciniphila may mediate these metabolic changes. It has been reported that A. muciniphila improves metabolism, inflammation, and outcomes of calorie restriction in obese patients [106107]. MBG may alter the enterohepatic circulation of bile acids, the gut microbiota, circulating bile acid composition, and bile acid pool size to improve metabolism in diabetes and obesity.
BILE ACID-BASED THERAPY FOR DIABETES
Bile acids as therapeutic drugs
Bile acids have been used directly to treat diabetes and obesity. Rectal taurocholate administration increased GLP-1 secretion from L-cells, insulin secretion from β-cells, and decreased serum glucose and food intake in diabetic patients [108]. TCA is converted to DCA, which activates intestinal FXR and TGR5 signaling to improve glucose and insulin tolerance. CDCA increased adipose tissue browning in humans [47], while UDCA was used to treat obese patients in a small cohort study of morbid obesity [109]. Short term UDCA administration stimulated bile acid synthesis and reduced circulation of FGF19 by inactivating FXR. Bile acid and cholesterol synthesis was enhanced, though serum and liver triglycerides were increased, and stearoyl-CoA was induced in white adipose tissues, generating less toxic monosaturated fatty acids. Metformin alters the gut microbiota to increase TUDCA and GUDCA, which antagonize intestinal FXR to improve hyperglycemia in diabetic patients [87].
Targeting FXR
Targeting the bile acid receptors FXR and TGR5 has therapeutic potential for treating metabolic liver diseases [21]. Obeticholic acid (OCA, 6α-ethyl-CDCA) is a semisynthetic bile acid that activates FXR with 30-fold greater efficacy than CDCA. OCA inhibits bile acid synthesis, improves liver function, and reduces liver inflammation in primary biliary cirrhosis [110]. OCA is effective in improving NASH scores in clinical trials and is a promising drug therapy for NASH [111]. OCA inhibition of bile acid synthesis also induced the gram-positive bacteria Firmicutes and alleviated NASH in humans [112].
Targeting TGR5
TGR5-selective agonists have been shown to improve glucose homeostasis and metabolic diseases [113114]. Activation of TGR5 by a semisynthetic bile acid, INT-777 (6α-ethyl-23(S)-methylcholic acid), reduces macrophage inflammation, lipid loading and atherosclerosis by inhibiting nuclear factor κB (NF-κB) and proinflammatory cytokine production in low-density lipoprotein (LDL) receptor deficient mice [115]. TGR5 reduces NF-κB activation of proinflammatory cytokine production and reduces inflammation [116]. The TGR5-selective agonist INT-777, and FXR and TGR5 dual agonist INT-767 (6α-ethyl-3α, 7α, 23-trihydroxy-24-nor-5β-cholan-23-sulfate, sodium salt) promotes adipose tissue browning in mice [50]. Activation of both FXR and TGR5 also promotes GLP-1 secretion, improves glucose and lipid metabolism and reverses hepatic steatosis, insulin resistance, and CVD [50117118119120]. However, activation of TGR5 stimulates gallbladder proliferation and deficiency of TGR5 protects against cholesterol gallstone disease in mice [48]. Still, the negative effect of TGR5 activation in gallbladder in humans has not been reported. Intestine-selective TGR5 agonists could be developed for treating inflammatory bowel disease and diabetes.
Bile acid sequestrants
Bile acid sequestrants bind bile acids in the intestine to prevent bile acid reabsorption, thus reducing the bile acid pool size. This results in reduced FGF19 and increased CYP7A1 gene transcription and hepatic bile acid synthesis [121]. Bile acid sequestrants may increase DCA in the colon to stimulate TGR5-mediated secretion of GLP-1 [122]. By increasing bile acid synthesis, bile acid sequestrants increase hepatic LDL-cholesterol uptake, reducing hypercholesterolemia, but increase serum triglycerides and cause dyslipidemia [123]. Cholestyramine and colestipol are classic bile acid sequestrants used to treat cholesterol gallstone disease and hypercholesterolemia in human patients, while colesevelam, a second-generation bile acid sequestrant, improves glycemic control in T2DM patients [121124125].
Targeting FGF19 and FGF21
Patients with metabolic syndrome and obesity have reduced circulating FGF19 levels but increased FGF21 [102]. FGF19 increases energy metabolism and metabolic rate, reduces weight and improves glucose tolerance and insulin sensitivity in diet-induced obese mice [126]. Engineered FGF19 without tumorigenic activity may be useful to treat diabetes and obesity [127128129]. Conversely, serum FGF21 is increased in obese, diabetic and NAFLD patients [130131]. FGF21 is a metabolic regulator that also affects nutrient preference in humans and may be used to treat metabolic diseases [64132133134]. DPP4 is increased in islets of T2DM patients and DDP4 inhibitor-based therapies have had moderate success in reducing glucose intolerance and insulin resistance [135], while GLP-1 receptor agonists have shown promise for the management of T2DM and reducing weight [136].
CONCLUSIONS
Emerging research in bile acid metabolism in the last three decades has contributed to identifying bile acids as endogenous ligands for FXR and TGR5 which mediate glucose, lipid, and energy metabolism and maintain whole body metabolic homeostasis. Overnutrition, high fat and high calorie diets, sleep disruption, drugs, and alcohol reshape gut microbiome to alter bile acid homeostasis and lead to dyslipidemia, hyperglycemia, insulin resistance and pathogenesis of diabetes, obesity, and related liver and heart diseases. Basic research in bile acid metabolism has contributed enormously to our current understanding of the molecular mechanisms and pathogenesis of liver-related metabolic diseases. However, most research has been focused on mouse models. Many of the results regarding FXR regulation of glucose, lipid, and energy metabolism are contradictory and remained to be resolved. Bile acid synthesis pathways are remarkably similar between mouse and human, but bile acid composition is very different. Thus, results from mouse studies cannot be extrapolated to humans without verification, and it is difficult to study bile acid metabolism in human subjects. Many metabolomic and microbiomic studies are limited to correlational analysis of serum and fecal samples between apparently healthy age- and sex-matched controls and patients diagnosed with diseases. Nevertheless, results from mouse studies have been translated to bile acid-based drugs targeting FXR, and to a lesser extent TGR5 for treating diabetes and obesity. Bile acid-based drugs are being developed for treating NASH, diabetes and obesity. However, bile acid-based FXR drugs cause pruritus and decrease serum high-density lipoprotein, and TGR5 drugs inhibit gallbladder emptying. Tissue selective FXR and TGR5 agonists and antagonists may be developed to circumvent the undesired side effects of drugs on other tissues. It is anticipated that bile acid-based drugs will be approved for treating diabetes, and treatments for NASH and fibrosis are expected in the near future.

 XML Download
XML Download