

 PDF
PDF ePub
ePub Citation
Citation Print
Print



The Sulwon Award for Scientific Achievement is the Korean Diabetes Association's highest scientific award and honors an individual who has excellently contributed to the progress in the field of diabetes and metabolism. The Sulwon Award is named after an emeritus professor, Eung Jin Kim, who founded Korean Diabetes Association. Prof. Ki-Up Lee received the 10th Sulwon Award at 31st Spring Congress of Korean Diabetes Association, May 3 to 5, 2018 at Gwangju, Korea.
INTRODUCTION
Low-grade inflammation in adipose tissue is considered a major contributing factor in the development of obesity-associated insulin resistance and cardiovascular disease [1]. Despite the well-established role in causing insulin resistance, the cause of adipose tissue inflammation is presently unsettled. High-fat diet (HFD) feeding was found to change gut microbiota and gut barrier function to initiate endotoxemia and insulin resistance in rodents [23]. From this, it has been suggested that lipopolysaccharide-induced activation of immune cells may be the primary lesion of adipose tissue inflammation and insulin resistance. Apart from this extrinsic immune system activation, however, there is a possibility that intrinsic changes in adipocytes are responsible for cell death, which triggers activation of inflammatory responses in adipose tissue. Thus, more than 90% of macrophages infiltrating the adipose tissue of obese animals and humans are arranged around dead adipocytes, forming characteristic crown-like structures [45].
Mitochondria generate cellular energy in the form of adenosine triphosphate through glucose and lipid metabolism, and produce many biosynthetic intermediates [67]. Conversely, mitochondrial dysfunction leads to oxidative stress, cell death, inflammation, and metabolic dysfunction [89]. Mitochondria are abundant in skeletal muscle, heart, brown adipocytes, and the liver. On the other hand, the role of mitochondria in white adipocytes has long been neglected because of their low abundance. However, recent evidence suggests that mitochondria are essential for maintaining metabolic homeostasis in white adipocytes [101112]. During the past decade, we investigated the roles of mitochondrial function in white adipocytes and found that mitochondrial function is essential for adiponectin synthesis [131415]. In this short review article, we proposed our new hypothesis that mitochondrial dysfunction in adipocytes may be a primary cause of adipose tissue inflammation and compared this with a prevailing concept that “adipose tissue hypoxia” may underlie adipose tissue dysfunction in obesity [161718].
ADIPONECTIN SYNTHESIS AND MITOCHONDRIAL FUNCTION IN ADIPOCYTES
It is now clear that the adipocyte is not a pure energy store but rather an endocrine gland playing an essential role in energy metabolism. Adipocytes secrete bioactive peptides or proteins, collectively named “adipokines.” Adiponectin is the most abundant adipokine synthesized from adipocytes that has many favorable effects on metabolism, including improvement of insulin sensitivity and reduction of atherosclerotic processes and systemic inflammation [19]. Interestingly, unlike other adipokines, plasma adiponectin levels are paradoxically lower in obese individuals than normal-weight individuals [20]. However, the cause of this paradox has remained unclear.
Adipocytes undergo two stages of maturation: differentiation and hypertrophy [21]. During the early stage of maturation (differentiation), adipocytes have higher levels of metabolic activities and adiponectin secretion, whereas hypertrophic cells lose most of their functional activities and adiponectin secretion [21]. Mitochondrial biogenesis is increased during adipocyte differentiation [22]. Therefore, we conducted a series of studies with the assumption that mitochondrial function is linked to adiponectin synthesis.
Peroxisome proliferator-activated receptor γ (PPAR-γ) agonist (thiazolidinediones [TZDs]) increases insulin sensitivity and plasma adiponectin levels [2324]. Interestingly, this action of TZDs was associated with an increase in the mitochondrial content in adipocytes [1325]. Nuclear respiratory factor 1 (NRF-1) is a key transcription factor that regulates mitochondrial biogenesis [26]. NRF-1 activates mitochondrial gene expression by up-regulating mitochondrial transcription factor A (mtTFA), which is essential for the replication and transcription of mitochondrial DNA (mtDNA). TZDs increased the expression levels of NRF-1 and mtTFA in 3T3L1 adipocytes, and siRNA against mtTFA reversed the TZD-induced increase in adiponectin synthesis and mtDNA content [13]. Adenovirus-mediated overexpression of NRF-1 in cultured 3T3L1 adipocytes increased mtTFA expression and mtDNA content, and these changes were accompanied by increased levels of adiponectin synthesis. Impaired mitochondrial function increased endoplasmic reticulum (ER) stress. Also, agents causing mitochondrial dysfunction or ER stress reduced adiponectin transcription via activation of c-Jun NH2-terminal kinase (JNK) and consequent induction of activating transcription factor 3 (ATF3). This study was the first to show that mitochondrial function is linked to adiponectin synthesis in adipocytes [13].
Among the known nitric oxide (NO) synthases, endothelial NO synthase (eNOS) plays an important role in the regulation of vascular tone and blood pressure [2728]. However, eNOS is also essential for mitochondrial biogenesis [29]. In our study, eNOS inactivation decreased adiponectin secretion in cultured adipocytes, and plasma adiponectin level and mitochondrial biogenesis in white adipose tissue were decreased in adult eNOS knockout (K/O) mice. Moreover, NO supply increased adiponectin synthesis and mitochondrial biogenesis in cultured adipocytes and eNOS K/O mice. These results suggested that proper function of eNOS in adipocytes is necessary for mitochondrial biogenesis and adiponectin synthesis [14].
Cushing's syndrome is a prototype of metabolic syndrome, characterized by central obesity, hypertension, insulin resistance, and glucose intolerance. Elevated levels of either endogenous or exogenous glucocorticoids cause this phenomenon. However, circulating cortisol levels are not always increased in subjects with metabolic syndrome, suggesting an intracellular mechanism that links glucocorticoid metabolism and metabolic syndrome [3031]. 11β-Hydroxysteroid dehydrogenase type 1 (11β-HSD1) converts inactive glucocorticoids into active glucocorticoids [32], and 11β-HSD1 activity is increased in adipose tissues of leptin-resistant Zucker obese rats [33]. Also, adipocyte-specific overexpression of 11β-HSD1 in transgenic mice produced typical features of metabolic syndrome [34]. In our study, 11β-HSD1 mRNA expression was increased, and mitochondrial respiration was decreased during prolonged culture of 3T3L1 adipocytes. Treatment with AZD 6925, a selective 11β-HSD1 inhibitor, increased mitochondrial biogenesis in cultured adipocytes. It also increased plasma adiponectin levels in db/db mice. This study may explain how hypertrophic adipocytes of obese subjects having decreased mitochondrial function produce a lower amount of adiponectin.
Taken together, it is suggested that mitochondrial function is essential for the production of adiponectin in adipocytes.
STUDIES SUPPORTING THE ROLE OF ADIPOCYTE MITOCHONDRIAL DYSFUNCTION IN INSULIN RESISTANCE
Consistent with our studies, other groups also reported that mitochondrial dysfunction in adipocytes may cause obesity or insulin resistance. Cellular levels of mitochondrial proteins, mitochondrial DNA contents, and mitochondrial function were decreased in obese (ob/ob) and diabetic (db/db) mice [1325]. More recently, an inverse regulation of inflammation and mitochondrial function was found in adipose tissue of morbidly obese patients [35]. Haplodeficiency of Crif1, a protein required for the intramitochondrial production of mtDNA-encoded oxidative phosphorylation (OXPHOS) subunits, reduced adipose OXPHOS capacity and triggered inflammation and insulin resistance in mice [36]. Mice deficient in mtTFA in adipocytes showed impaired mitochondrial biogenesis in adipose tissue, lipodystrophic syndrome with insulin resistance, hepatosteatosis, and cardiovascular complications [37]. In humans, mtDNA contents, mtDNA-encoded transcripts, and mitochondrial OXPHOS protein levels in adipose tissue were downregulated in the obese compared with the lean co-twins [38]. Pathway analysis indicated downshifting of fatty acid oxidation, ketone body production and breakdown, and the tricarboxylic acid cycle, which was inversely correlated with adiposity, insulin resistance, and inflammatory cytokines [38].
Taken together, these results suggested that mitochondrial dysfunction is found in adipocytes of human and animals with obesity, and that this is related to insulin resistance, adipose tissue inflammation, and/or lipodystrophy.
ADIPOCYTE MITOCHONDRIAL DYSFUNCTION IN AGING
Increasing evidence suggests that mitochondrial function is a potential central regulator of the aging process [39]. Mitochondrial dysfunction contributes to cellular senescence, chronic inflammation, and the age-dependent decline in stem cell activity [40]. A recent study reported that mitochondrial complex IV dysfunction is pivotal in aging-associated obesity [41]. Plasma adiponectin levels are decreased in apparently healthy elderly men and women [42], and aging is associated with circulating inflammatory markers independent of body mass index [43]. Taken together, these results suggest that mitochondrial dysfunction in adipocytes is related to adipose tissue inflammation in aging as well as obesity or insulin resistance.
HYPOTHESIS: MITOCHONDRIAL DYSFUNCTION MAY BE A PRIMARY CAUSE OF ADIPOSE TISSUE INFLAMMATION
Immune cell infiltration is a prominent feature of dysfunctional adipose tissue, and these immune cells include M1 and M2 macrophages, effector and memory T-cells, interleukin 10 producing FoxP3+ T regulatory cells, natural killer and natural killer T (NKT) cells, and granulocytes [44]. Of these various immune cells, adipose tissue macrophages play a central role in the genesis of adipose tissue inflammation [45]. At least two distinct populations of macrophages infiltrate the adipose tissue: proinflammatory (M1) and anti-inflammatory (M2). Among the M1 and M2 macrophage markers, inducible nitric oxide synthase (iNOS) and arginase are considered the most important molecules responsible for M1 and M2 activities, respectively [45]. iNOS metabolizes arginine to NO and citrulline. On the other hand, arginase expressed in M2 macrophages hydrolyzes arginine to ornithine and urea.
Macrophages have been shown to aggregate, forming crown-like structures surrounding necrotic adipocytes in obesity [446]. Therefore, it was proposed that necrosis of adipocytes is a prominent phagocytic stimulus that activates adipose tissue macrophage infiltration [4]. Mitochondria play a central role in cell death [4748]. Thus, mitochondrial dysfunction in adipocytes may trigger cell death in adipocytes to induce adipose tissue inflammation.
Monocyte chemoattractant protein-1 (MCP-1) is produced predominantly by macrophages and endothelial cells and is a potent chemotactic factor for monocytes. However, it is now well recognized that adipocytes are an important source of MCP-1 production [49]. mRNA expression in adipose tissue and the plasma concentration of MCP-1 were increased in obese mice [50]. Recently, mitochondrial dysfunction was shown to increase MCP-1 expression in renal tubular epithelial cells causing inflammation [51]. Thus, there is a possibility that mitochondrial dysfunction in adipocytes causes increased expression of MCP-1 in adipocytes to increase chemotaxis of macrophages into adipose tissue.
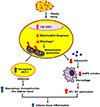
Finally, adiponectin is well-known to activate AMP-activated protein kinase (AMPK) in diverse tissues [52], and adiponectin activates AMPK in macrophages [53]. AMPK shifts M1 to M2 macrophage polarization [5455], and adiponectin was shown to prime human monocytes into anti-inflammatory M2 macrophages [56]. Therefore, we propose that failure to produce adiponectin synthesis due to mitochondrial dysfunction in adipocytes may lead to decreased M2 macrophage polarization and increased adipose tissue inflammation (Fig. 1).
PROINFLAMMATORY RESPONSE OF MACROPHAGES MAY EXACERBATE MITOCHONDRIAL DYSFUNCTION IN ADIPOCYTES
As described above, mitochondrial dysfunction in adipocytes may be the primary mechanism that triggers adipose tissue inflammation and insulin resistance. However, proinflammatory cytokines were shown to regulate adipocyte mitochondrial metabolism [57]. We thus examined the other possibility that proinflammatory response of macrophages induces mitochondrial dysfunction in adipocytes.
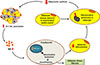
In addition to inflammation, adipose tissue fibrosis is exacerbated in obese human subjects and HFD-fed rodents. Fibrosis limits the expandability of adipose tissue and contributes to ectopic fat accumulation and the development of insulin resistance [45]. Classically activated (M1) macrophages have increased iNOS expression and contribute to adipose tissue inflammation. We examined the relationship between NO produced by macrophages and adipose tissue fibrosis. The hypoxia-inducible factor 1α (HIF-1α) protein level was increased in adipose tissue of wild-type (WT) mice fed an HFD for a prolonged period but not in iNOS K/O mice. The expression of mitochondrial biogenesis factors was decreased in HFD-fed WT mice but not in iNOS K/O mice. In studies with cultured cells, macrophage-derived NO decreased the expression of mitochondrial biogenesis factors and increased DNA damage and phosphorylated p53 in preadipocytes. By activating p53 signaling, NO suppressed expression of peroxisome proliferator-activated receptor gamma coactivator 1α (PGC-1α), which induced mitochondrial dysfunction and inhibited preadipocyte differentiation to adipocytes. The effects of NO were blocked by rosiglitazone. These findings suggest that NO produced by macrophages induces mitochondrial dysfunction in preadipocytes by activating p53 signaling, which in turn increases HIF-1α protein levels and promotes a profibrogenic response in preadipocytes that results in adipose tissue fibrosis [45].
These results suggest that, in addition to primary mitochondrial dysfunction in adipocytes, there may be additional mechanisms that promote and sustain mitochondrial dysfunction in adipocytes and/or adipose tissue inflammation/fibrosis (Fig. 2).
MITOCHONDRIAL QUALITY CONTROL AND MITOPHAGY
To ensure adequate function of mitochondria, quality control mechanisms are necessary to eliminate damaged mitochondrial proteins or parts of the mitochondrial network by mitophagy and renew components by adding protein and lipids through biogenesis [58]. When dysfunctional mitochondria are not cleared adequately by mitophagy, this may lead to aging-associated diseases [59].
In mammals, mitophagy is mediated either by the PTEN-induced putative kinase 1 (PINK1)-Parkin signaling pathway or the mitophagic receptors Bnip3 and Nix [60]. Even though the role of mitophagy in adipocytes has not been studied adequately, a recent study found that PINK1-Parkin alleviates metabolic stress induced by obesity in adipose tissue and 3T3-L1 preadipocytes [61].
CLINICAL IMPLICATION
Then, what would be the clinical implication of these findings? Exercise training has long been known to increase mitochondrial biogenesis in skeletal muscle [62]. In addition, recent studies found that exercise training increases mitochondrial biogenesis in white adipose tissue [6364]. Thus, the beneficial effect of exercise training may, at least in part, be due to its effect on mitochondria in adipose tissue.
TZDs are known to increase mitochondrial biogenesis in adipocytes [65] and are currently used in clinical medicine to treat diabetes mellitus and insulin resistance. There may be many other chemical or natural compounds that improve mitochondrial function [66] in adipocytes through increase of mitochondrial biogenesis and/or mitophagy, but their role in clinical medicine remains to be determined.
COMPARISON OF OUR HYPOTHESIS WITH HYPOXIA THEORY
Finally, we would like to compare our hypothesis with a prevailing concept that “adipose tissue hypoxia” may underlie adipose tissue dysfunction in obesity. In the year 2007, two groups of researchers proposed that adipose tissue of obese mice is hypoxic, and that local adipose tissue hypoxia dysregulates the production of adiponectin [1667]. Since then, this has been regarded as the main mechanism of insulin resistance by many investigators [6869]. However, we found that mitochondrial dysfunction in preadipocytes of HFD-fed mice induced pseudo-hypoxic induction of HIF-1α protein levels, which led to adipose tissue fibrosis [45].
In fact, cellular response to hypoxia and pseudo-hypoxic response by mitochondrial dysfunction is very similar in that both conditions are characterized by decreased mitochondrial respiration and increased glycolysis. “Adipose tissue hypoxia” theory explains that there is hypoxia in enlarged adipocytes distant from the vasculature [70]. The only method that could determine whether adipose tissue hypoxia is present in adipose tissue of obesity is the measurement of oxygen tension. However, the results are quite controversial. Most studies performed in rodents reported that adipose tissue oxygen tension is decreased in obese animals [166771]. On the other hand, a study performed in humans reported increased adipose tissue oxygen tension despite lower adipose tissue blood flow in obese compared with lean individuals [72]. This was accompanied by insulin resistance, lower adipose tissue capillarization, lower adipose tissue expression of genes encoding proteins involved in mitochondrial biogenesis and function, and higher macrophage infiltration and inflammatory markers.
CONCLUSIONS
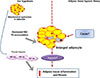
In this article, we proposed that mitochondrial dysfunction in adipocytes may be a primary cause of adipose tissue inflammation (Fig. 1). Despite widespread belief on adipose tissue hypoxia theory, we hypothesize that mitochondrial dysfunction in adipocytes is the primary cause of insulin resistance and adipose tissue inflammation (Fig. 3). Defective mitochondrial function and decreased fatty acid oxidation in adipocytes would increase triglyceride accumulation to cause adipocyte enlargement and adipose tissue hypoxia. In addition, mitochondrial dysfunction in adipocytes may exert a “pseudohypoxic response” to promote adipose tissue fibrosis, making it difficult to distinguish these two theories from each other. Even though adipose tissue hypoxia has been commonly observed in mouse obesity, human data reported increased adipose tissue oxygen tension [60]. In addition, mitochondrial dysfunction in adipose tissue was found in human obesity [3761], supporting our hypothesis that mitochondrial dysfunction in adipocytes is a primary cause of insulin resistance in obesity. The cause of mitochondrial dysfunction in adipocytes in obesity is presently unknown, but future studies are warranted to test whether a defective mitochondrial quality control mechanism is responsible for mitochondrial dysfunction in hypertrophic adipocytes.
 XML Download
XML Download