

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Prolonged exposure of β-cells to elevated levels of free fatty acids (FFAs) leads to decreased glucose-stimulated insulin secretion (GSIS) and apoptosis [12]. Several mechanisms underlying β-cell lipotoxicity have been proposed, such as endoplasmic reticulum (ER) stress, mitochondrial dysfunction, reactive oxygen species (ROS) production, and islet inflammation [345]. The molecular mechanisms triggered by elevated FFA seem to act interdependently to create a vicious cycle that leads to β-cell failure.
It is well known that metformin stimulates AMP-activated protein kinase (AMPK). AMPK inactivates acetyl-CoA carboxylase (ACC) which plays an essential role in regulating fatty acid synthesis and oxidation [6]. It has been reported that the AMPK-dependent pathway was required for the lipid-lowering and insulin-sensitizing effects of metformin in the liver [78]. Metformin is also known to directly inhibit complex I on the mitochondrial respiratory chain of isolated liver mitochondria at millimolar concentrations [9]. This action of metformin has been shown to inhibit hepatic glucose production by decreasing the hepatic energy state even in the absence of AMPK activation [10]. A recent study reported that the glucose-lowering effect of metformin was accompanied by a marked and instant change in the cytosolic and mitochondrial redox state of hepatocytes. This action of metformin was attributed to the inhibition of the mitochondrial redox shuttle, glycerophosphate dehydrogenase, and is achieved by relatively low concentrations that were comparable to plasma concentrations observed in patients treated with metformin [11].
There have been few studies that have investigated the effects of metformin in pancreatic β-cells. Some studies have reported that metformin restores insulin secretion of cultured islets impaired by chronic exposure to elevated FFA [1213]. In contrast, others have reported that metformin resulted in impaired GSIS and apoptosis of β-cells through AMPK stimulation [14]. In one study, metformin was shown to play a dual role in β-cell apoptosis depending on the culture condition [15]. The effects of metformin on β-cell function, whether protective or damaging, seem to be partly attributed to the difference in concentration of metformin used between experiments.
This study was motivated by the observation of a relationship between the action of metformin and its concentration. In the present study, we aimed to ascertain the effects of metformin on lipotoxicity-induced β-cell dysfunction. In particular, we focus on the relation of metformin to the dependence on AMPK as well as the concentration of metformin in examining its action on β-cell lipotoxicity as metformin concentration in the pancreas is expected to be low compared to the liver concentration, and was close to the concentration observed in plasma of patients treated with metformin at usual clinical doses (20 mg/kg), which was known to be in the range of 0.01 to 0.04 mM [16].
METHODS
Cell culture and reagents
NIT-1 cells were purchased from ATCC (Cat# CRL-2055, RRID: CVCL_3561; Manassas, VA, USA) and cultured in RPMI 1640 medium (Gibco Life Technologies, Carlsbad, CA, USA) supplemented with 10% (v/v) fetal bovine serum (FBS; Gibco Life Technologies), 100 U/mL penicillin, and 100 µg/mL streptomycin. Palmitate was dissolved at 100 mM in ethanol to make stock solution. Palmitate stock solution was incubated at 37℃ for an hour and diluted in the culture medium to which fatty acid-free bovine serum albumin (BSA) had been added in a 1:3 molar ratio, to prepare BSA-conjugated palmitate. Cells were incubated with the BSA-conjugated palmitate (0.2 to 0.5 mM) for the indicated time periods. The compounds 1,1-dimethylbiguanide hydrochloride (metformin) and 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) used as an AMPK agonist as well as compound C as an AMPK antagonist were purchased from Sigma (St. Louis, MO, USA).
Cell viability assay
Cell viability was assessed using Cell Counting Kit-8 (Dojindo Molecular Technologies, Rockville, MD, USA). Fifty microliters of cell counting kit-8 (CCK-8) solution was added to 500 µL of culture medium. Cells were incubated for 30 minutes at 37℃ and then the absorbance was measured at 450 nm using a microplate reader. The net absorbance from the plates of cells with the control medium was considered as 100% of cell viability.
Measurement of GSIS
NIT-1 cells were seeded in a 6-well plate at 104 cells/well in RPMI 1640 containing 10% FBS, after which 200 µM palmitate was added to the medium for 48 hours. The cells were starved for 2 hours in glucose-free RPMI 1640, washed twice with a glucose-free 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)-balanced Krebs-Ringer bicarbonate buffer (KRBH) containing 0.2% fatty acid-free BSA, and pre-incubated for 1 hour in the same medium. After pre-incubation, the cells were exposed to KRBH buffer containing 2.5 mM (low glucose) or 16.7 mM (high glucose) glucose for 1 hour. Insulin secreted into the supernatant was measured by the insulin immunoassay kit (ALPCO, Salem, NH, USA) and was adjusted for total cellular protein of cell lysate. The measurement from the plate of cells with the control medium stimulated by low glucose was considered as 100% of GSIS.
Isolation of mouse pancreatic islets and measurements of GSIS
Female C57BL/6J and BALB/c mice at 24 to 33 weeks of age were used as islet donors. The animal use protocol of this study was approved by the Institutional Animal Care and Use Committee of Seoul National University (IACUC No. SNU-150327-3-2). The isolation of islets from mice was carried out according to a previously described protocol [17]. The isolated islets that were with 100 to 200 µm in size were picked into culture plate (15 to 20 per well) and incubated at 37℃ in culture medium (RPMI 1640 supplemented with 10% FBS, 100 U/mL penicillin, and 0.1 mg/mL streptomycin) for 3 hours. After which, islets were treated with 0.5 mM palmitate and metformin for 24 hours. After the islets were incubated for 20 minutes in low-glucose (2.5 mM) KRBH buffer, the islets medium was replaced with fresh low-glucose KRBH. After 40 minutes of exposure to the low glucose, the medium was collected, and islets were incubated with high-glucose (16.7 mM) KRBH buffer for 40 minutes. At the end of the incubation, the medium was collected. Insulin was measured by the insulin immunoassay kit (ALPCO) according to the manufacturer's instructions.
RNA extraction and real-time quantitative polymerase chain reaction
Total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. The cDNA was prepared by reverse transcription with 1 µg of total RNA. Real-time polymerase chain reaction (PCR) was performed using SYBR Premix Ex Taq reagents (TaKaRa, Shiga, Japan) and a 7500 real-time PCR system (Applied Biosystems, Foster City, CA, USA). The glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA level was used for the internal control. Experiments were performed in duplicate for each sample and repeated four times. The following primer sequences were used: activating transcription factor 4 (ATF4), 5′-CCTGAACAGCGAAGTGTTGG-3′ (forward), 5′-TGGAGAACCCATGAGGTTTCAA-3′ (reverse); C/EBP homologous protein (CHOP), 5′-CAACAGAGGTCACACGCACA-3′ (forward), 5′-TCTCCTTCATGCGTTGCTTC-3′ (reverse); glucose-regulated protein 94 (GRP94), 5′-AAACGGCAACACTTCGGTCAG-3′ (forward), 5′-GCATCCATCTCTTCTCCCTCATC-3′ (reverse); FK506 binding protein 11 (FKBP11), 5′-ACACGCTCCACATACACTACACGG-3′ (forward), 5′-ATGACTGCTCTTCGCTTCTCTCCC-3′ (reverse); NADPH oxidase (NOX) 1, 5′-AATGCCCAGGATCGAGGT-3′ (forward), 5′-GATGGAAGCAAAGGGAGTGA-3′ (reverse); NOX2, 5′-CCCTTTGGTACAGCCAGTGAAGAT-3′ (forward), 5′-CAATCCCGGCTCCCACTAACA-3′ (reverse). Relative mRNA levels were expressed as a fold-change relative to the vehicle-treated control.
Small interfering RNA transfection in NIT-1 cells
The small interfering RNAs (siRNAs) for mouse Rho kinase 1 (ROCK1) and AMPK were synthesized as Stealth siRNA duplexes (Invitrogen). The siRNA of the negative control was purchased from Bioneer (Seoul, Korea). The siRNA was incubated with lipofectamine RNAiMAX reagent (Invitrogen) in 100 µL serum-free medium for 15 minutes. The siRNA-Lipofectamine RNAiMAX complex was then added to the cells in 400 µL of serum-containing medium and maintained for 1 day, and then the cells were treated with palmitate and metformin as described.
Measurement of intracellular ROS level
The cells were incubated with 2′, 7′-dichlorohydro-fluorescein diacetate (DCF-DA; Invitrogen) in RPMI 1640 supplemented with 10% FBS for 30 minutes. Fluorescence was measured with an excitation at 450 nm and emission at 535 nm using a Victor 3 1420 multilabel counter (PerkinElmer, Boston, MA, USA). The measurement from the plate of cells with the control medium was considered as 100% of intracellular ROS level.
ROCK1 activity assay
NIT-1 cell lysates (300 µg protein) were subjected to immunoprecipitation with polyclonal ROCK1 antibody (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), coupled to protein G-Sepharose beads (PharmaciaBiotechnology, Piscataway, NJ, USA). Immune pellets were washed and resuspended in 50 µL of kinase mixture (20 mM Tris [pH 7.5], 5 mM MgCl2, 100 mM KCl, 0.1 mM dithiothreitol, 100 M adenosine triphosphate [ATP], 1 mM ethylenediaminetetraacetic acid [EDTA], 1 M microcystin-LR, 50 M long S6K substrate peptide [Millipore, Bedford, MA, USA], and 1 Ci [-32P] ATP) and incubated at 30℃ for 30 minutes. ROCK1 activity was measured as described previously [18].
Measurement of intracellular adenosine diphosphate/ATP ratio
For determination of the adenosine diphosphate (ADP)/ATP ratio, the ADP/ATP Ratio Assay Kit (Abcam, Cambridge, UK) was used according to manufacturer's instructions. Cells seeded in a 24-well plate and treated were lysed with nucleotide-releasing reagent that had been supplied in the kit and 50 µL of the lysate was transferred to a 96-well plate. To measure ATP levels, the luminescence was measured using a Victor 3 1420 multilabel counter (PerkinElmer) after 100 µL of the substrate solution, which was also supplied, was added to each well. To measure ADP levels in the cells, the luminescence was measured again after 10 µL of ADP converting enzyme was added to each well.
Western blot analysis
Cells were washed with phosphate buffered saline, harvested, and incubated in lysis buffer containing 20 mM HEPES (pH 7.4), 1% Triton X-100, 15% glycerol, 2 mM ethylene glycol tetraacetic acid (EGTA), 1 mM sodium vanadate, 2 mM dithiothreitol, 10 µM leupeptin, and 5 µM pepstain. Total protein extracts (30 µg) were separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE). Proteins on the gel were transferred onto a nitrocellulose membrane. Membranes were incubated with blocking solution (5% skim milk), and then incubated with primary antibodies against phosphorylated AMPK and total AMPK (Cat# 2531, RRID: AB_330330 and Cat# 2532, RRID:AB_330331; Cell Signal Technology, Danvers, MA, USA) in 0.1% Tween 20-Tris-buffered saline. Hybridized primary antibodies were detected using a horseradish peroxidase-conjugated immunoglobulin G antibody (Cat# sc-2357, RRID:AB_628497; Santa Cruz Biotechnology). Bands were detected by using an enhanced chemiluminescence kit (Thermo, Rockford, IL, USA).
RESULTS
Metformin restores cell viability and GSIS impaired by palmitate
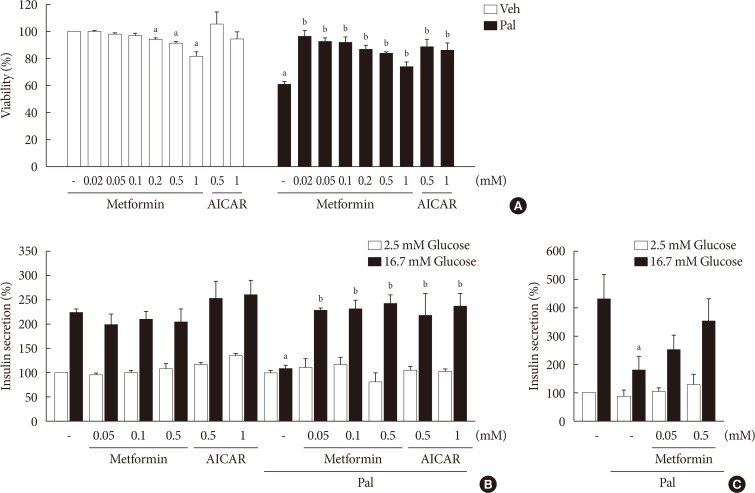
When NIT-1 cells were treated with palmitate (0.5 mM) for 24 hours, the viability was decreased significantly. Metformin inhibited palmitate-induced death of NIT-1 cells over a concentration range of 0.02 to 1.0 mM. AICAR (0.5 to 1.0 mM), an AMPK activator, also inhibited cell death induced by treatment with palmitate (Fig. 1A). In NIT-1 cells, GSIS was nearly abolished by treatment with palmitate (0.2 mM) for 48 hours. Metformin restored GSIS impaired by treatment with palmitate over a concentration range of 0.05 to 0.5 mM. AICAR exerted a similar effect on GSIS impaired by palmitate treatment (Fig. 1B). GSIS was reduced by about 60% in islets exposed to 0.5 mM palmitate for 24 hours. Metformin partially restored GSIS impaired by treatment with palmitate at 0.05 and 0.5 mM in mouse islets (Fig. 1C).
Lower metformin concentration (0.05 mM) suppresses palmitate-induced elevation of intracellular ROS level and NOX and the expression of ER stress markers
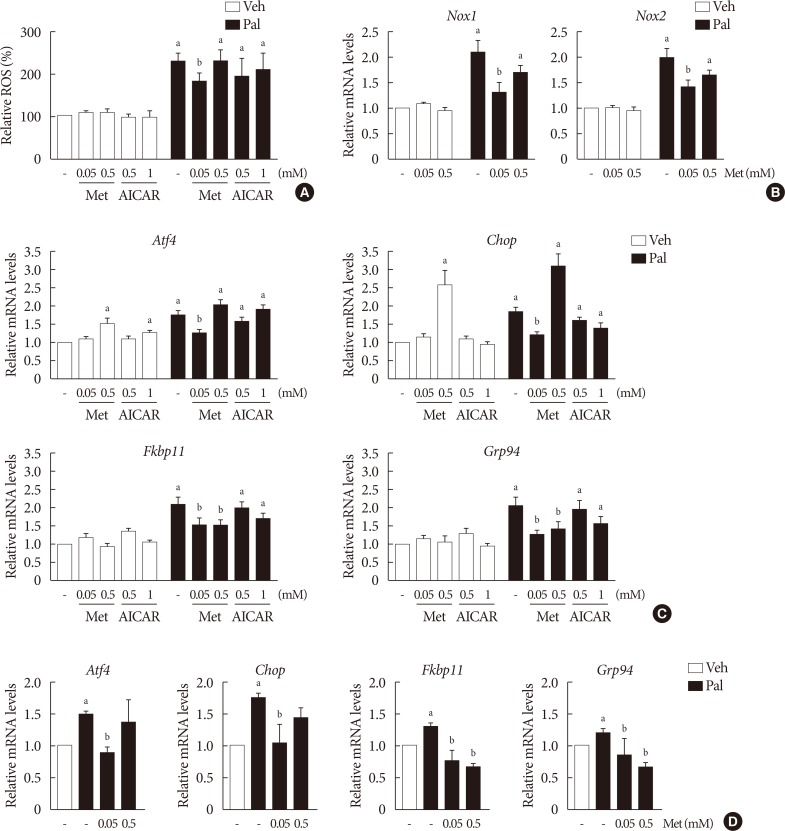
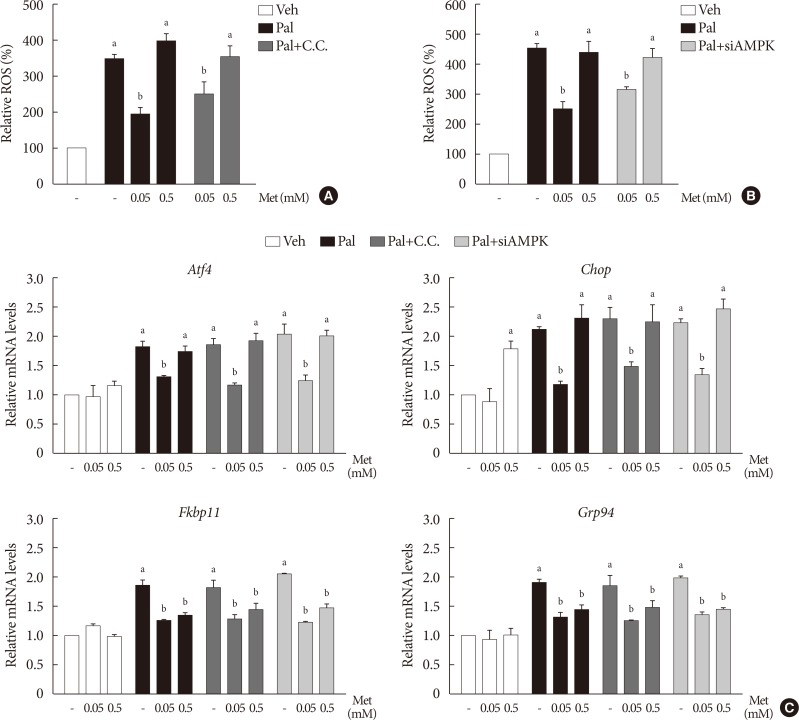
Treatment with palmitate (0.5 mM) for 24 hours triggered a 2.0- to 2.5-fold increase of intracellular ROS fluorescence in NIT-1 cells. Metformin at a concentration of 0.05 mM inhibited the ROS increment due to palmitate, while 0.5 mM metformin and AICAR (0.5 to 1.0 mM) had little effect on intracellular ROS (Fig. 2A). Treatment with palmitate (0.5 mM) for 24 hours triggered a 1.5- to 2.0-fold increase of mRNA levels of NOX1/NOX2 in NIT-1 cells. Metformin at a concentration of 0.5 mM had no effect on the expression levels of NOX1 and NOX2 that were increased by the treatment with palmitate, whereas 0.05 mM metformin inhibited palmitate-induced NOX1/2 expressions (Fig. 2B). In NIT-1 cells, the mRNA levels of ATF4, CHOP, FKBP11, and GRP94 increased by 1.5- to 2.0-fold by treatment with palmitate (0.5 mM) for 24 hours. Low concentrations of metformin (0.05 mM) consistently reduced ER stress markers induced by palmitate treatment, while 0.5 mM metformin reduced FKBP11 and GRP94, but not ATF4 or CHOP. The effects of AICAR (0.5 to 1.0 mM) on the expression levels of these ER stress markers were not significant (Fig. 2C). We observed similar effects of metformin on the expression levels of these ER stress markers in isolated mouse islets exposed to palmitate (Fig. 2D).
Higher metformin concentration (0.5 mM) increases the intracellular ADP/ATP ratio and AMPK phosphorylation
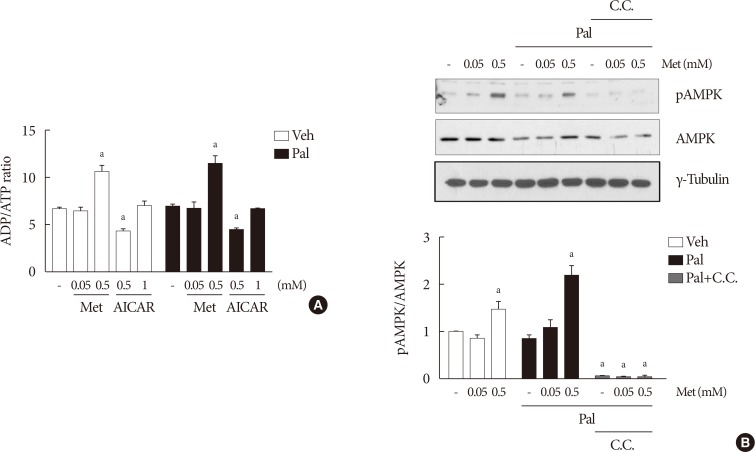
The cellular ADP/ATP ratio was not altered by treatment with palmiate (0.5 mM) for 24 hours in NIT-1 cells. Metformin at a concentration of 0.5 mM resulted in an increase in the cellular ADP/ATP ratio (Fig. 3A) and levels of AMPK phosphorylation (Fig. 3B) in NIT-1 cells regardless of exposure to palmitate. Metformin at a concentration of 0.05 mM resulted in neither an increase in the cellular ADP/ATP ratio nor an increase in the levels of AMPK phosphorylation in NIT-1 cells exposed to palmitate. Levels of AMPK phosphorylation were nearly nullified by compound C, an AMPK antagonist, when it was added with metformin (Fig. 3B).
The effects of metformin on cellular ROS levels and ER stress markers elevated by palmitate are not affected by inhibition of AMPK
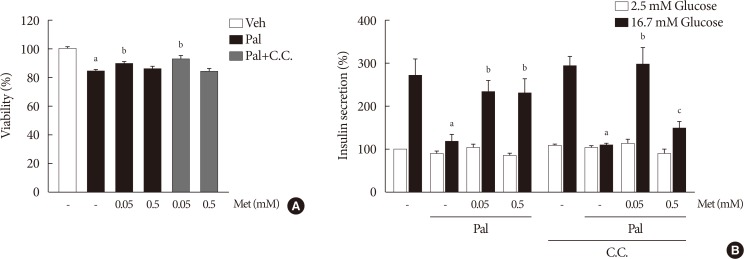
When compound C (0.01 mM), an AMPK inhibitor, was added to the metformin treatment in NIT-1 cells exposed to palmitate, intracellular ROS levels were not altered (Fig. 5A). The siRNAs for AMPK (100 nM) also had no effects on intracellular ROS levels when added to the metformin treatment in NIT-1 cells exposed to palmitate (Fig. 5B). Likewise, inhibition of AMPK by compound C and siRNAs for AMPK did not alter the effect of metformin on the expression levels of ER stress markers (ATF4, CHOP, FKBP11, GRP94) in NIT-1 cells exposed to palmitate (Fig. 5C).
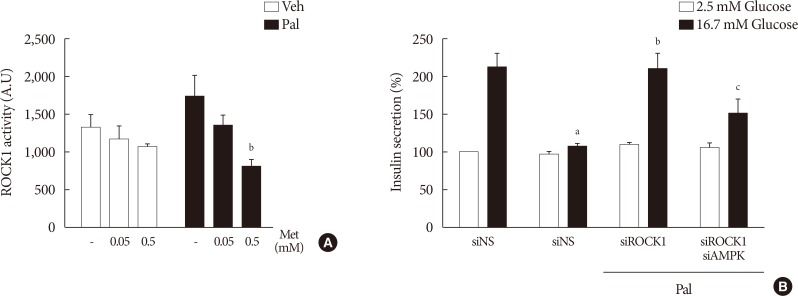
Metformin at a concentration of 0.5 mM inhibits ROCK1 in NIT-1 cells exposed to palmitate, and ROCK1 inhibition restores GSIS impaired by palmitate
Metformin at a concentration of 0.5 mM inhibited ROCK1 activity in NIT-1 cells exposed to palmitate (Fig. 6A). The siRNAs for ROCK1 (100 nM) restored GSIS impaired by palmitate treatment (Fig. 6B). The siRNAs for AMPK (100 nM) significantly attenuated the effect of ROCK1 inhibition to improve GSIS (Fig. 6B).
DISCUSSION
Our results showed that metformin restored viability and GSIS of β-cells exposed to palmitate independently of AMPK at a low concentration (0.05 mM), but was dependent on AMPK at a high concentration (0.5 mM). It is thus supposed, that the action of metformin on pancreatic β-cells was implemented by different pathways depending on its concentration. At lower concentrations, metformin inhibited NOX expression, alleviated lipotoxicity-induced oxidative stress and ER stress, and consequently improved β-cell function in the presence of lipotoxic stress. This pathway altered neither cellular energy state nor the activation of AMPK. At higher concentrations, metformin stimulated AMPK by depleting cellular energy charge, inhibited ROCK and improved lipotoxic β-cell dysfunction without affecting oxidative stress. This AMPK-dependent pathway may account for a dual action of metformin in β-cells, which trigger or alleviate β-cell dysfunction according to the absence or presence of lipotoxic stress.
ER stress signaling is involved at least partially in lipoapoptosis of β-cells [19]. In a study on the lipotoxicity of clonal β-cells, metformin showed a protective activity against lipotoxic apoptosis through attenuation of ER stress at 0.01 to 0.1 mM, which was consistent with the results of this study [20]. On the contrary, in another study, 5 mM metformin ameliorated thapsigargin-induced dysfunction of clonal β-cells through the AMPK pathway, but failed to block ER stress markers [21]. These findings suggest that the action of metformin on ER stress was sensitive to its concentration and to a type of ER stress inducer in pancreatic β-cells. In primary rat cardiomyocytes, metformin was shown to induce specific pathways of ER stress signaling leading to strong and persistent induction of CHOP at concentrations higher than 2 mM [22]. In spite of CHOP induction, metformin treatment did not trigger apoptosis, which was also observed in our study in the absence of lipotoxic stress. In this study, the effects on ER stress markers of 0.5 mM metformin were different between ATF4/CHOP and FKBP11/GRP94. The results from previous studies suggest that activation of ATF4/CHOP pathway by 0.5 mM metformin might be attributed to mitochondrial complex I inhibition induced by metformin at this concentration [2324].
Oxidative stress is another mechanism underlying β-cell lipotoxicity. It has been reported that ROS levels are dose- and time-dependently increased by exposure of NIT-1 cells to palmitate and that suppression of NOX2 restores FFA-induced dysfunction of NIT-1 cells [25]. There is an evidence that a positive feedback cycle exists between oxidative stress and ER stress in β-cell dysfunction and this cycle can be interrupted by superoxide dismutase mimetics and chemical chaperones [26]. In other cell types, it has been demonstrated that both protein misfolding in the ER and ROS are required to activate cellular responses, thereby causing cell death, and antioxidant treatment can reduce ER stress to improve cell survival and protein secretion [27]. Furthermore, there is also evidence demonstrating that ER stress can induce oxidative damage in pancreatic β-cell, though the exact mechanism for the generation of ROS has not been elucidated [2829]. In this study, lower concentration metformin inhibited NOX expression and ROS production that were induced by 0.5 mM palmitate. It was likely that lower concentration metformin disrupted the reciprocal link between ER stress and oxidative stress to improve β-cell dysfunction induced by lipotoxicity. In contrast, higher concentration metformin improved lipotoxic β-cell dysfunction without having any effects on ROS production. The protective effects of higher concentration metformin can be partly attributed to the direct action on mitochondria to limit metabolism as a previous finding suggested that dysregulation of mitochondrial metabolism is the primary cause of lipotoxicity occurring upstream of ROS accumulation [30].
AMPK controls metabolic homeostasis by acting as a cellular energy sensor [31]. However, its role in pancreatic β-cell, especially in β-cell dysfunction under lipotoxic condition, remains controversial [32]. In the present study, exposure to palmitate had little effect on the cellular ADP/ATP ratio and phosphorylation level of AMPK in spite of definite negative impacts on β-cell function, suggesting that AMPK per se does not play a major role in β-cell lipotoxicity. However, AICAR, a specific activator of AMPK, restored β-cell viability and glucose-induced insulin secretion impaired by lipotoxicity. Metformin also exerted its effects on lipotoxic β-cell dysfunction in an AMPK-dependent manner at concentrations at which activation of AMPK occurs. On the contrary, ER stress and oxidative stress induced by lipotoxicity remained unaffected by the activation or inhibition of AMPK. Since AICAR and metformin have been claimed to reduce hepatic lipid accumulation through AMPK [83334], and accumulation of detrimental lipids is known to be an initiator of β-cell failure [3536], it is natural to assume that downstream targets of AMPK that regulate intracellular lipids would mediate the AMPK-dependent effects of metformin on β-cell lipotoxicity. However, according to previous findings, metformin had no effect on triglyceride overload in β-cells despite its protective effect on lipotoxicity [1337], highlighting the difference in AMPK-dependent action of metformin between the liver and pancreas. Taken together, AMPK in β-cells mediates the effects of metformin at concentrations equal to or higher than 0.5 mM in the presence of lipotoxic stress via an unknown downstream target, which seems unrelated to known mechanisms underlying β-cell lipotoxicity and hepatic insulin resistance.
The ROCK signaling pathway is known to mediate insulin resistance in the muscle induced by elevated FFA levels [38]. Metformin has been reported to exert its protective effects against vascular dysfunction by decreasing ROCK activity [39]. In this study, we found that 0.5 mM metformin suppressed ROCK1 activity in NIT-1 cells exposed to palmitate, and that inhibition of ROCK1 activity reproduced the effects of 0.5 mM metformin on GSIS impairment induced by lipotoxicity through AMPK activation. A previous study reported that inhibition of ROCK ameliorated metabolic disorders through the activation of the AMPK pathway in the skeletal muscle and liver [40]. Based on these findings, it is supposed that ROCK1 is a possible target candidate for the action of 0.5 mM metformin which is dependent of AMPK activation, although the exact molecular interactions between metformin and ROCK1 in pancreatic β-cells remains to be elucidated.
It has been reported that intracellular ATP content is decreased by metformin at concentrations greater than 0.25 mM with concomitant stimulation of AMPK in primary hepatocytes [610]. In contrast, plasma concentrations of metformin at usual clinical doses (20 mg/kg) have been shown to be in the range of 0.01 to 0.04 mM [16]. As metformin accumulates to higher levels in hepatocytes than in plasma due to the high expressions of organic cationic transporter 1 (OCT1), which facilitates cellular uptake of metformin, there will be a difference in tissue metformin concentration between the liver and other target organs including the pancreas. Therefore, we hypothesized that metformin at lower concentrations was able to improve lipotoxic β-cell dysfunction through inhibition of oxidative stress and ER stress, while metformin at higher concentrations reduces glucose output and lipogenesis in the liver through the activation of AMPK and depletion of cellular energy charge. A study in which plasma and tissue metformin concentrations were measured in rats treated with metformin reported that acute metformin administration led to metformin concentration in the pancreas of approximately 0.05 to 0.06 mM, which is lower than in the liver and comparable to lower concentrations of metformin used in this study [11]. Hence, if our findings hold true in in vivo setting, they are considered to be clinically relevant because the action of metformin presented in this study will be a possible explanation for the protective role of lower metformin concentration in β-cell lipotoxicity and for the difference in drug action at therapeutic doses between the liver and pancreas regarding the treatment of diabetes.
In conclusion, we demonstrated that metformin ameliorated palmitate-induced β-cell dysfunction through the inhibition of ER stress and cellular ROS production at concentrations achieved by a usual therapeutic dose in an AMPK-independent manner, which was in contrast with the effects of metformin at higher concentrations. NOX and ROCK are considered to be molecular targets for the concentration-dependent dual action of metformin in β-cell lipotoxicity. The findings from this study suggest that metformin exerts its action on lipotoxicity associated with diabetes through a different molecular mechanism between different target organs and different working concentrations.
 XML Download
XML Download