

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Diabetic kidney disease (DKD) is the most common microvascular complication of diabetes and accounts for the majority of cases of end-stage renal disease (ESRD) [1]. The most common predisposing factor of ESRD is tubulointerstitial injury, mediated by processes such as interstitial fibrosis and inflammation [2]. Accumulating evidence demonstrates that mononuclear inflammatory cells are involved in damaging the renal interstitium and that persistent inflammation contributes to tubulointerstitial fibrosis [34]. Thus, understanding the mechanisms regulating inflammation in ESRD is likely to aid development of an effective therapeutic strategy for preventing tubulointerstitial fibrosis.
Dipeptidyl peptidase-4 (DPP-4) inhibitors (also known as gliptins) have proven protective effects on DKD. Their glucose lowering effects are mediated through decreasing the degradation of glucagon-like peptide 1 (GLP-1) [5]. As DPP-4 is localized on the surface of many cell types, including endothelial cells, kidney epithelial cells, and T-cells, many studies have demonstrated that DPP-4 inhibitors have pleiotropic effects in various organ systems [6]. In terms of expression per weight of organ, the kidney bears the highest levels of DPP-4 and, as DPP-4 induces inflammation, the protective effect of gliptins on renal fibrosis have been extensively investigated [6]. Linagliptin was found to ameliorate kidney fibrosis in streptozotocin-induced diabetic mice [7], and the renoprotective effect of alogliptin was demonstrated in mice with unilateral ureteral obstruction (UUO)-induced renal interstitial fibrosis [8]. Previously, we also demonstrated that gemigliptin plays a role in the prevention of DKD, regardless of its glucose-lowering effect [9].
The inflammasomes are a group of cytosolic protein complexes that function as immune system receptors and sensors in response to microbial infection and cellular damage [10]. Among the NOD-like receptors (NLRs), which sense whole pathogens or toxins and regulate the immune response, NOD2 and NOD-like receptor protein 3 (NLRP3) are the best understood [11]. The NLRP3 inflammasome and its initiation of production of proinflammatory cytokines have been investigated in many inflammatory settings including atherosclerosis, rheumatic diseases, microbial infection, and chronic inflammatory diseases [121314]. Upon activation, the NLRP3 proteins oligomerize and recruit the adaptor protein apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), and caspase-1, which assemble to form the inflammasome, and which leads to the conversion of the cytokine precursors pro-interleukin 1β (pro-IL-1β) and pro-IL-18 into biologically active IL-1β and IL-18, respectively [15]. Recently, growing evidence has revealed that the inflammasome significantly contributes to the progression of chronic kidney disease (CKD), including both tubulointerstitial and glomerular disease [12]. The NLRP3 inflammasome is activated in the UUO-induced tubular interstitial inflammation model of renal fibrosis (UUO kidney), whereas Nlrp3-deficient mice have less tubular injury and inflammation compared with the wild type [3]. However, the effect of gemigliptin on NLRP3 inflammasome activation in a model of renal fibrosis has not been fully investigated.
Here, we investigated whether the anti-fibrotic effect of gemigliptin in the UUO kidney is related to changes in NLRP3 inflammasome activity. In addition, we evaluated the effect of gemigliptin on the transforming growth factor-β (TGF-β)/nuclear factor-κB (NF-κB) signaling pathway, known to be a primary pathogenic factor in renal fibrosis and a potential mechanism underlying the effect of gemigliptin, in vivo and in vitro.
METHODS
Experimental design
For UUO-induced renal fibrosis, after a midabdominal incision under anesthesia using pentobarbital (50 mg/kg), the left ureter of C57BL6 mice was ligated with 5-0 silk suture at two separate points and cut between the two ligation points. After UUO, mice were orally administered 300 mg/kg/day gemigliptin. The dose of gemigliptin used in this study was determined by preciously published studies [91617]. Fourteen days after UUO and gemigliptin treatment, mice were euthanized, and their left kidneys were removed, cut in thirds, fixed in 4% paraformaldehyde, and either embedded in paraffin for histologic examination or frozen in liquid nitrogen for the isolation of protein.
Histologic and morphologic analysis
Kidneys were fixed by immersion in phosphate-buffered saline containing 4% paraformaldehyde overnight and then embedded in paraffin. Sections (4 µm) were cut and deparaffinized in xylene, followed by rehydration in a graded series of ethanol. Staining was performed using hematoxylin and eosin (H&E), and Sirius red. Immunohistochemical (IHC) staining was performed using primary antibodies against the fibronectin (BD Biosciences, San Jose, CA, USA), plasminogen activator inhibitor 1 (PAI-1; BD Biosciences), and type I collagen (Abcam, Cambridge, UK), and NLRP3 (Novus Biologicals, Littleton, CO, USA), ASC, caspase-1, IL-1β (Santa Cruz Biotechnology, Santa Cruz, CA, USA), CD-26 (Abcam), and NF-κB (Cell Signaling Technology, Danvers, MA, USA), followed by horseradish peroxidase-conjugated anti-mouse or anti-rabbit immunoglobulin G secondary antibodies (Dako, Glostrup, Denmark), according to the manufacturer's instructions. Renal fibrotic areas were quantitated by morphometric analysis using an MRc5 Carl Zeiss light microscope (Oberkochen, Germany) equipped with an imaging system and iSolution DT version 7.7 software (IMT i-Solution, Coquitlam, BC, Canada). Sirius red-positive areas and immunostaining for fibronectin, type I collagen, PAI-1, NRLP3, ASC, Caspase-1, and IL-1β in the renal fibrotic regions (brown color) were quantitated by computer-based morphometric analysis. All data were normalized to the control and expressed as fold increase relative to the control.
Cell culture
Human kidney proximal tubular epithelial human renal proximal tubule cells (HK-2) cells were purchased from the American Type Culture Collection (Manassas, VA, USA), and cultured in 5% CO2 at 37℃ in keratinocyte growth medium (GIBCO, Carlsbad, CA, USA) containing 0.2 ng/mL human recombinant epidermal growth factor and 50 µg/mL bovine pituitary extract. Cells were treated with gemigliptin (200 or 400 µg/mL) in culture medium with or without TGF-β (5 ng/mL; Sigma, St. Louis, MO, USA) for 48 hours. Cells were subsequently processed for the isolation of protein as described below.
Western blot analysis
For protein preparation, cells and kidney tissue were suspended in radioimmunoprecipitation assay buffer. The cells were then lysed on ice for 30 minutes, and the cell lysate was collected by centrifugation at 15,000×g for 10 minutes. Protein quantitation was performed using a Bio-Rad Protein Assay kit (Bio-Rad, Richmond, CA, USA). Then, 30 µg of proteins were electrophoresed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and electrotransferred to polyvinylidene fluoride membranes (Millipore, Bedford, MA, USA). After blocking with 5% skimmed milk in Tris-buffered saline containing Tween 20 (0.1%) for 1 hour, the membrane was incubated with anti-fibronectin (1:1,000; BD Biosciences), anti-PAI-1 (1:1,000; BD Biosciences), anti-type I collagen (1:1,000; Abcam), anti-α smooth muscle actin (α-SMA) (1:1,000; Sigma), anti-NLRP3 (1:1,000; Novus Biologicals), anti-ASC (1:1,000; Santa Cruz Biotechnology), anti-caspase-1 (1:1,000; Santa Cruz Biotechnology), anti-IL-1β (1:1,000; Santa Cruz Biotechnology), and anti-NF-κB (1:1,000; Cell Signaling Technology) polyclonal antibodies at 4℃ with gentle shaking overnight. Expression was detected by horseradish peroxidase-linked secondary antibody (Santa Cruz Biotechnology) using the enhanced chemiluminescence Western Blotting Detection system, according to the manufacturer's instructions (Amersham, Buckinghamshire, UK). The membrane was reblotted with anti-β-tubulin antibody to verify equal loading of the protein in each lane. Densitometric measurements of the bands were made using the UN-SCAN-IT digitizing program (Silk Scientific Corp., Orem, UT, USA).
Statistical analysis
Data were evaluated using analysis of variance followed by a post hoc least significant difference test and expressed as mean±standard error of mean. Values of P<0.05 were considered statistically significant. All experiments were performed at least three times.
Ethical statements
All procedures were performed in accordance with institutional guidelines for animal research [18]. This article does not contain examinations performed on human participants. Then, ethical approval was not necessary.
RESULTS
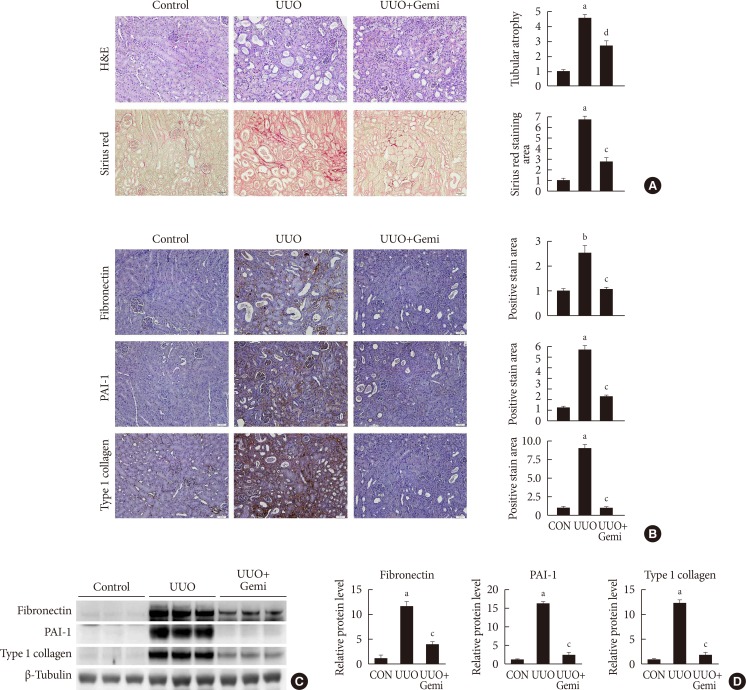
Gemigliptin ameliorates UUO-induced renal fibrosis
First, we examined the effect of gemigliptin on UUO-induced renal fibrosis. H&E and Sirius red staining showed that UUO-induced tubular atrophy and tubulointerstitial fibrosis were significantly attenuated by treatment with gemigliptin (Fig. 1A). IHC staining demonstrated that gemigliptin reduced UUO-induced expression of fibronectin, PAI-1, and type I collagen (Fig. 1B). The effect of gemigliptin on UUO-induced expression of fibrogenic proteins was additionally confirmed by Western blot analysis (Fig. 1C and D).
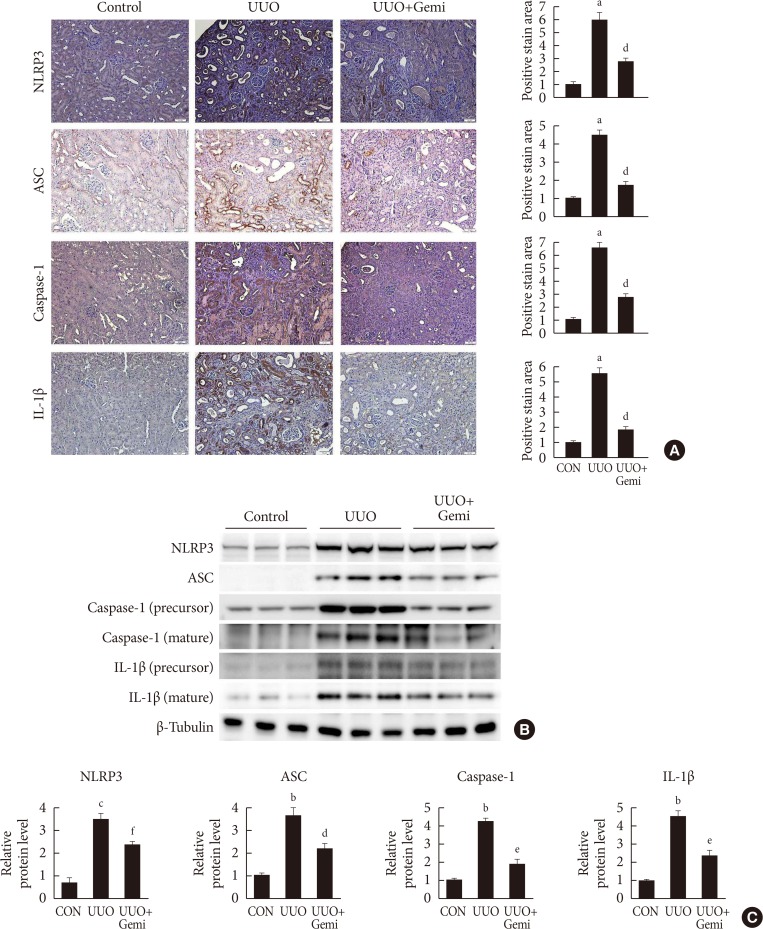
Gemigliptin attenuates the UUO-induced NLRP3 inflammasome
To investigate the effect of gemigliptin on NLRP3 inflammasome activation in renal fibrosis, we analyzed the levels of NLRP3 inflammasome components NLRP3, ASC, caspase-1, and mature IL-1β in UUO kidneys. IHC staining revealed that the levels of NLRP3, ASC, caspase-1, and IL-1β were increased after ureteral obstruction. By contrast, activation of the NLRP3 inflammasome in UUO kidneys was inhibited by gemigliptin treatment (Fig. 2A). Consistent with the IHC staining results, as determined by Western blot, the protein levels of NLRP3, ASC, caspase-1, and mature IL-1β were lower in gemigliptin-treated UUO kidneys than in vehicle-treated kidneys (Fig. 2B and C).
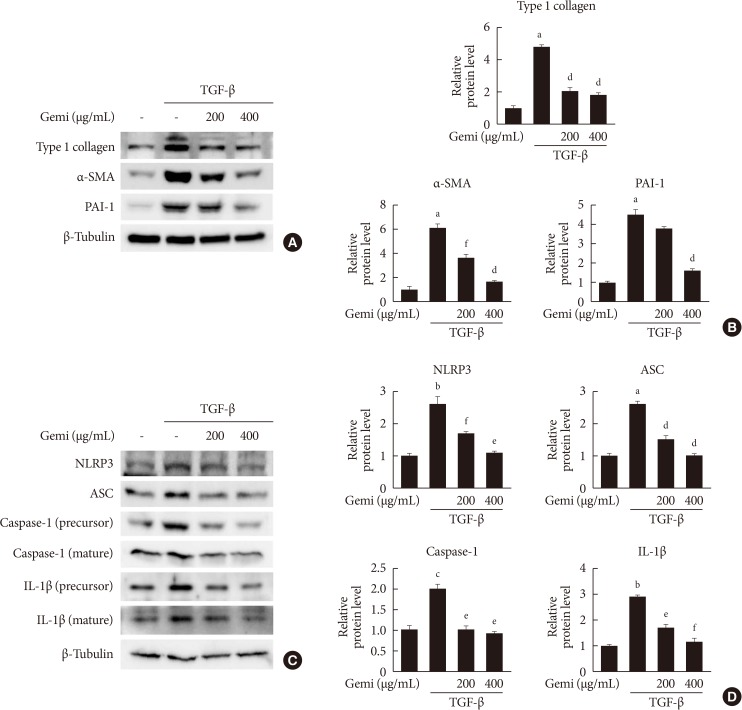
Gemigliptin inhibits expression of TGF-β-stimulated profibrotic- and NLRP3 inflammasome-related proteins
Given that TGF-β is a primary pathogenic factor in renal fibrosis [19], we evaluated whether gemigliptin had an effect on the level of TGF-β-induced profibrotic proteins in cultured renal cells. As expected, TGF-β treatment increased protein levels of type I collagen, α-SMA, and PAI-1 in HK-2 cells. Gemigliptin-treated HK-2 cells showed markedly inhibited TGF-β-stimulated profibrotic protein level in a dose-dependent manner (Fig. 3A and B). Moreover, gemigliptin inhibited TGF-β-induced upregulation of NLRP3, ASC, caspase-1, and IL-1β in a dose-dependent manner (Fig. 3C and D). These results suggest that gemigliptin has a renoprotective effect by suppressing the activation of the NLRP3 inflammasome.
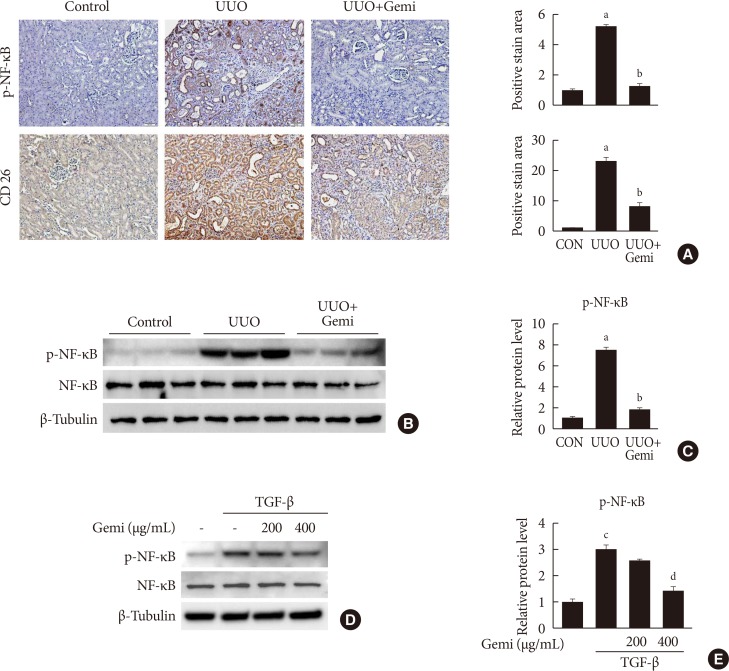
Gemigliptin inhibits NF-κB activation in vivo and in vitro
To evaluate the mechanism by which gemigliptin attenuates UUO-induced extracellular matrix (ECM) accumulation and NLRP3 inflammasome activation, we examined whether NF-κB activation (phosphorylated NF-κB) was induced in vivo by UUO, or in vitro by TGF-β treatment of HK-2 cells, and whether gemigliptin inhibited its activation. Furthermore, to assess the contributions of local DPP-4 on UUO-induced renal fibrosis, we also examined renal DPP-4 levels. IHC staining showed areas of staining of phosphorylated NF-κB and DPP-4 were increased in sections of UUO kidney, but these activation was significantly reduced by gemigliptin treatment (Fig. 4A). This inhibitory effect of gemigliptin on NF-κB activation was further confirmed by Western blot analysis (Fig. 4B and C). In accordance with the in vivo findings, gemigliptin effectively inhibited TGF-β-stimulated NF-κB phosphorylation in HK-2 cells in a dose-dependent manner (Fig. 4D and E).
DISCUSSION
In the present study, we demonstrated that gemigliptin had an apparent preventive effect on renal fibrosis. This study showed that gemigliptin abrogated UUO-induced ECM accumulation. Furthermore, gemigliptin inhibited NLRP3 inflammasome activation and subsequent proinflammatory cytokine production in the UUO kidney. Collectively, the renoprotective effect of gemigliptin appears to the consequence of down-regulated fibrotic gene expression via the suppression of TGF-β/NF-κB-induced NLRP3 inflammasome activation.
There is increasing evidence that examination of NLRP3 inflammasome activation offers new insights into the variable pathogenesis of renal fibrosis [20]. In a mouse model of crystal nephropathy, intratubular crystal formation triggers intrarenal inflammasome activation, which can be abrogated by a NLRP3-specific inflammasome inhibitor [21], while in a mouse model of type 1 and 2 diabetes mellitus, increased levels of NLRP3, ASC1, and caspase-1 demonstrate NLRP3 inflammasome activation in the diabetic kidney, which can be inhibited by saxagliptin halting progression of nephropathy [22]. Furthermore, it is known that IL-1β, which is produced by the activation of the NLRP3 inflammasome, plays an important role in development and progression of renal fibrosis [23]. Consistent with previous results, the present study also revealed that NLRP3 inflammasome activation and inflammasome-dependent IL-1β production is increased in UUO-induced fibrotic kidneys, and suppressed in the kidneys of gemigliptin-treated mice. In accordance with the in vivo finding, gemigliptin inhibited TGF-β-stimulated ECM proteins as well as inflammasome-related proteins in cultured renal tubular cells.
The proinflammatory transcription factor NF-κB regulates multiple aspects of innate and adaptive immune responses [24]. In the context of the inflammasome, NF-κB is critical for the priming signal of NLRP3 inflammasome activation and functions by inducing the transcriptional regulation of NLRP3 and pro-IL-1β in response to inflammatory signals [25]. Considering that TGF-β is a major player in renal fibrosis and it activates NF-κB signaling, targeting NF-κB could lead to amelioration of TGF-β-induced renal fibrosis [2627]. Previous reports identified that the anti-inflammatory function of DPP-4 inhibitors is at least in part mediated by NF-κB inhibition [28]. In the present study, we found that phosphorylation of NF-κB was increased in the UUO-induced fibrotic kidney and in TGF-β-stimulated renal cells. Treatment with gemigliptin abrogated NF-κB activation, which provided evidence that the renoprotective effect of gemigliptin on renal fibrosis via the NLRP3 inflammasome was mediated by down-regulation of NF-κB signaling.
Our results agree with those of previous studies, which demonstrated that DPP-4 inhibition attenuates upregulation of DPP-4 immunolabeling in the fibrotic kidney compared with control kidney [729]. Previous reports demonstrated that exogenous TGF-β treatment enhanced DPP-4 activity in proximal tubule cells [29] and local DPP-expression increases inflammation by binding to caveolinn-1 and activating NF-κB [30]. Therefore, our data support the possibility that enhanced DPP-4 levels contribute to NF-κB activation and subsequently priming NLRP3 inflammasome in UUO-induced fibrotic kidney. However, we cannot exclude other possible mechanisms responsible for the renoprotective effect of gemigliptin such as regulation of GLP-1R expression or substrate including GLP-1, stromal cell-derived factor 1α (SDF-1α) [313233]. Considering that administration of gemigliptin increased serum GLP-1 level in UUO mice (data not shown), GLP-1 may mediate some of the beneficial effect of gemigliptin. Further investigation would be required for confirming direct contribution of DPP-4 on inflammasome and renal fibrosis.
In summary, we demonstrated that gemigliptin has a renoprotective effect on renal fibrosis by regulation of the NLRP3 inflammasome. Inhibition of the NLRP3 inflammasome is the target for the prevention of renal fibrosis progression. Therefore, the present study provides the rationale for further study to elucidate the effect of gemigliptin in inhibition of the NLRP3 inflammasome in the clinical setting for preventing CKD as well as DKD.
 XML Download
XML Download