

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
The mammalian immune response can be roughly divided into two categories. The type 1 immune response is an acute, highly inflammatory program intended to activate phagocytic cells, such as neutrophils, T helper type 1 (Th1) cells, and type 1 macrophages (M1) to destroy the targets, and is often associated with tissue damage [1]. The type 1 immune response is strongly facilitated by interleukin 2 (IL-2), interferon γ (IFN-γ), and lymphotoxin-α secreted by Th1 lymphocytes. In contrast, the type 2 immune response is a more long-lived program that plays a major role in barrier defenses, such as intestinal motility and mucus barriers, and tissue remodeling, including fibrosis and wound healing [2]. The type 2 immune response is characterized by T helper 2 (Th2) cells, eosinophils, mast cells, basophils, type 2 innate lymphoid cells (ILC2s), IL-4 and/or IL-13-induced macrophages, immunoglobulin E (IgE), and the cytokines IL-4, IL-5, IL-9, and IL-13 [23]. When the Th1–Th2 dichotomy was first described [4], type 2 immunity was generally considered a regulatory function, not as an important response to pathological conditions, and its primary function was to limit the injurious consequences of type 1-mediated immunity [5]. Although this original limited definition remains accurate, recent studies have found that the role of type 2 immunity includes acting as a major effector response that has many important host-protective and pathogenic activities, in addition to suppressing type 1 immunity and type 1-driven inflammation [6]. In addition, these cell populations also show protective activity by reducing inflammation in the process of tissue regeneration [7].
Many recent studies have focused on the immune regulation of adipose tissue (AT) to achieve systemic glucose homeostasis in metabolic disease. AT can be broadly divided into two categories: brown adipose tissue (BAT), which is a highly catabolic tissue that undergoes high levels of thermogenesis, and white adipose tissue (WAT), an anabolic tissue that serves as a primary long-term nutrient storage site [89]. AT is comprised of stromal and vascular cells, including innate and adaptive immune cells, fibroblasts, and endothelial cells in addition to adipocytes, and recent research has focused on immune mediators as a major axis of metabolic regulation [101112]. Previously, AT in the obese state was shown to promote inflammation mediated by type-1 signals, including infiltration and expansion of M1 macrophages with secretion of inflammatory cytokines, such as tumor necrosis factor-α (TNF-α), IL-6, and IL-1β. However, more recent studies have identified the pivotal role of type 2 immunity, including regulatory T (Treg) cells, Th2 cells, and ILC2 cells, which suppress inflammatory responses by production of the anti-inflammatory cytokine, IL-10, and contribute to the development of insulin resistance [13141516]. There is emerging evidence that the IL-33–driven ILC2/eosinophil axis plays a role in browning of WAT, and eosinophils in AT are involved in metabolic homeostasis via IL-4/IL-13–mediated reconstitution of macrophages, thereby preventing development of obesity [1718]. Although there has been pioneering work on the regulatory role of the ILC2 population in AT, such as the recovery of ILC2 in obesity by injection of IL-33 in obese mice [19], the mechanisms underlying the loss of ILC2 by metabolic stress have not been identified [20]. Although ILC2-derived cytokines have been shown to induce browning of WAT, how ILC2-derived cytokines affect systemic glucose metabolism is not well understood [2122].
In this review, we discuss how the type 2 immune response and Th2 cytokines regulate metabolism in AT, its most well-studied context. Specifically, we review the systemic regulatory roles of Th2 cytokines in metabolic disease and the role of mitochondria in maintenance of type 2 responses in AT homeostasis. We also briefly discuss the pathological consequences of their dysfunction.
ROLE OF TYPE 2 IMMUNE CELLS IN ADIPOSE TISSUE HOMEOSTASIS
Recent observations have shown that obesity results in an inflammatory microenvironment in AT, leading to the deterioration of tissue-protective mechanisms that reduce the negative outcomes of immunopathology. Although AT in lean mice and humans contains a higher proportion of immune cells associated with type 2 immunity, type 2 immune cells are reduced in number during the development of obesity [2324]. Recent studies have suggested the importance of ILC2s as central regulators of type 2 immunity in an anti-inflammatory microenvironment in AT [2526]. However, the regulatory effects of ILC2-derived Th2 cytokines with systemic metabolic regulation are not fully understood. In this section, we review the role of type 2 immune cells, including anti-inflammatory macrophages, ILC2, and eosinophils, in physiological AT homeostasis.
M2 macrophages
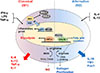
Adipose tissue macrophages (ATMs) are abundant immune cells in human and murine AT [2728]. ATMs, which have substantial functional heterogeneity, actively participate in various aspects of nutrient metabolism within AT [29]. Macrophages identified in lean AT are anti-inflammatory “M2”, or alternatively activated (M2-like) macrophages, which typically express the mannose receptor CD206 and secrete anti-inflammatory cytokines (Fig. 1) [3031]. AT in lean mice and humans contains a higher proportion of M2/M1 macrophages, which are associated with local production of Th2 cytokines by eosinophils [1832]. Initially, the involvement of the immune response in AT homeostasis was understood only in the context of type 1 immunity, represented by the increased proinflammatory M1 macrophages and production of IFN-γ and chemokines, such as CCL2 released from CD8+ and Th1-polarized lymphocytes, as well as their ability to exacerbate metabolic dysfunctions, such as type 2 diabetes mellitus and obesity [33].
M2 macrophages play a role in nutrient storage in adipocytes by promoting insulin sensitivity through the secretion of IL-10 [34], and suppress type 1 immune responses of leukocyte populations by processing excess iron, disposing of dead cells and debris, and promoting vascularization and tissue matrix remodeling [3536]. The regulatory key mediators of macrophage polarization were recently identified [37]. M1 macrophages have huge metabolic demands depending on glycolysis [3839]. M2 macrophages meet their energy requirements using mitochondrial oxidative phosphorylation (OXPHOS) pathways, and have increased ability to uptake free fatty acid and for fatty acid oxidation (FAO) to facilitate the tricarboxylic acid cycle (TCA) cycle, and drive inflammasome activation in inflammatory macrophages [3740]. Inhibition of FAO in M2 macrophages with the carnitine palmitoyltransferase 1 (CPT1) inhibitor, etomoxir, limits M2 activation in response to IL-4 [34]. Peroxisome proliferator-activated receptor γ/δ (PPAR-γ/δ), which can be driven by adipocyte derived IL-4 and IL-13, is a crucial transcription factor in M2 macrophage polarization [4142]. In contrast, M1 macrophages increase energy production through glycolysis. M1 macrophages also have a “broken” TCA cycle, resulting in accumulation of citrate and succinate. Increased succinate results in secretion of the proinflammatory cytokine, IL-1β, via activation of hypoxia-inducible factor 1α (HIF1α) (Fig. 1) [404344]. Blocking oxidative metabolism not only impairs the development of an M2-like phenotype but also drives the cells toward an M1-like state [45]. In addition, failure of alternative M2 activation, which is associated with reduced oxidative function, leads to classical macrophage activation, elevated weight gain, and obesity with concurrent adipose inflammation and insulin resistance [4647]. However, it is unclear whether the oxidative function of macrophages is reduced under these conditions, and it is not known whether treatments capable of increasing the oxidative function of macrophages would reverse insulin resistance and adipose inflammation.
ILC2
ILC2s are a recently identified innate immune cell lineage related to lymphocytes and natural killer cells with emerging roles in mediating immune responses and regulating tissue homeostasis and inflammation [48]. The group 2 ILC population is comprised of ILC2s, which express IL-5 and IL-13 and require GATA-binding protein 3 (GATA3) and retinoic acid receptor-related orphan receptor α (ROR-α) for their development [49]. In a mouse model of intestinal damage and inflammation, the epithelial cytokine IL-33 can promote ILC2 amphiregulin (Areg) production, leading to the resolution of colitis and promoting epithelial repair [50]. Indeed, type 2 immune responses are known to promote wound repair and tissue regeneration following infection or injury [1], suggesting that ILC2s may be key organizers of these beneficial tissue responses. Although they have crucial roles in protective type 2 immunity, the role of these cells in WAT is quite distinct [51]. They function to promote WAT homeostasis through several defined mechanisms.
ILC2s are present in visceral adipose tissue (VAT), where they are the predominant producers of IL-5 and IL-13 following prolonged exposure to IL-33 [2552]. In lean AT, IL-33 drives the recruitment and/or proliferation of ILC2, but the cellular origin of IL-33 and the mechanisms leading to its secretion in homeostasis remain poorly understood. Tissue ILC2s are key producers of systemic IL-5 required for homeostatic eosinophil maintenance [53]. In AT, secretion of IL-5 by ILC2 is essential for the recruitment and maintenance of eosinophils and is dependent on IL-33 [53]. In addition, the most well-studied mechanism involves their regulatory effect on the resident macrophage phenotype from M1 macrophages to M2 macrophages by IL-4 or IL-13 signaling [54]. The regulatory function of these Th2 cytokines is involved in systemic glucose homeostasis. IL-4 and IL-13, both induced by type 2 immune responses, reduce inflammation in AT and improve systemic glucose intolerance by inducing polarization of M2 macrophages [185556]. Interestingly IL-33 has been shown to be competent to induce macrophage proliferation independently of IL-4Ra expression in other non-adipose macrophage populations [53], but whether IL-33 can directly activate ATMs remains to be investigated.
Eosinophils
Eosinophils are a type of granulocyte that combat parasitic infection and allergic reactions. However, in AT their role is to maintain metabolic homeostasis [18]. Initial studies in WAT showed that eosinophils are associated with lean healthy WAT and promote M2-like polarization of WAT macrophages [1825]. WAT eosinophils decline with weight gain in association with increased AT M1-like macrophages and metabolic impairments, and eosinophil-deficient mice are more vulnerable to the onset of insulin resistance [18]. Further research has shown that WAT eosinophil numbers are maintained by IL-5, which is largely secreted by local ILC2s [1825]. One study showed that body weight and glucose control are improved by ILC2 and natural killer T cells in a model of high fat diet-induced obesity, which was attributed to the accumulation of eosinophils and M2-like macrophages in visceral WAT [57]. Recently, it was found that resident eosinophils expressing IL-4 and ILC2s expressing IL-13 are necessary to maintain an anti-inflammatory state in WAT [1825]. During cold exposure, eosinophils may induce WAT beiging by polarizing M2-like macrophages [5859]. These results suggest that ILC2s, eosinophils, and M2-like macrophages influence metabolic health through the regulation of AT inflammation, beiging capacity, and insulin resistance by facilitating a network of immune cell interactions (Fig. 1). In the simplest terms, AT M2-like macrophages, eosinophils, and ILC2s are positively correlated with AT health.
Treg cells
Treg cells are abundant in the visceral AT of lean mice, where they have a distinct transcriptome and antigen-receptor repertoire [60]. Similar to ILC2s, Treg cells rapidly populate AT after birth. AT Treg cells express high levels of CD25 and IL-10, as well as the transcription factors GATA3 and PPAR-γ, and the IL-33 receptor [15]. They uniquely depend on these molecules, since mice lacking IL-33, IL-33R, or PPAR-γ expression in Treg cells are deficient in Treg cells specifically within AT and exhibit evidence of increased AT inflammation [3252]. In addition, IL-33-driven expansion of ILC2s and/or Treg cells has been shown to revert the chronic inflammatory processes that drive obesity and improve insulin resistance in mouse models [61].
THE LOCAL EFFECTS OF TH2 CYTOKINES, ILC2, AND M2 MACROPHAGES IN ADIPOSE TISSUE INFLAMMATION
Recent findings support a regulatory role for type 2 immunity in AT and systemic metabolism, although the precise mechanisms remain to be determined. The possible mechanisms for the effects of type 2 immunity on AT metabolism include local effects, such as AT browning, AT remodeling, and limiting excessive type 1 inflammatory response, and systemic effects through the production of adipocytokines. In this section, we discuss the local effects of type 2 immunity in AT inflammation.
Obesity is associated with local adipose inflammation, which is characterized by dysregulated immune cell function [62]. CD8+ T cells play a critical role in high-fat diet-induced obesity and adipose inflammation via macrophage recruitment and activation [63]. ATMs secrete inflammatory cytokines, such as TNF-α and IL-6. Secretion of TNF-α, plasminogen activator inhibitor-1, IL-6, IL-1β, and other inflammatory cytokines by ATs is higher in obese patients than in lean individuals [6465]. As discussed below in the section on AT homeostasis, emerging evidence suggests that obesity is related to weakening of the type 2 immune response, since transitioning of the macrophage phenotype is likely a key mechanism by which IL-4 and/or IL-13 protect against metabolic syndrome. Accordingly, other pathways that promote M2 macrophages are also associated with metabolic homeostasis, such as the fatty acid-sensing PPAR-γ/PPAR-δ, adenosine receptor 2B, and Krüppel-like factor 4 (KLF4) pathways [666768]. Macrophage-specific deletion of PPAR-γ or PPAR-δ increases obesity, decreases glucose tolerance, and increases insulin resistance in mice fed high-fat diets [46]. Consequently, oxidative functions of ATMs control the polarization of M1-like and M2-like phenotypes, but whether reduced macrophage oxidative function causes systemic insulin resistance in vivo is not fully understood.
Recent studies have suggested a role for IL-4/IL-13–mediated reconstitution of macrophages by eosinophils in AT [18]; furthermore, administration of recombinant IL-4 to mice also reduces weight gain and has a protective effect against diet-induced obesity [55]. Importantly, IL-4 improves the metabolic indices of insulin resistance and improves the action of insulin in the liver and muscle in a signal transducer and activator of transcription 6 (STAT6)-dependent manner [55]. Although the increase in WAT eosinophils by recombinant IL-5 treatment is not related to lipid and glucose metabolism, IL-33/IL-25 upregulates WAT ILC2, which recruits eosinophils via IL-5, resulting in increased M2 macrophages and improved WAT homeostasis [57], suggesting a pivotal role for ILC2 in glucose homeostasis [69]. In addition, cold or exogenous IL-33 stimulates ILC2 and eosinophil production of IL-13 and IL-4, respectively, which drive the proliferation and commitment of adipocyte precursors to beige fat [5970]. IL-33-activated ILC2s produce methionine-enkephalin peptides, which act directly on adipocytes to stimulate beige adipogenesis independently of eosinophils [17]. These results suggest that type 2 immunity has a regulatory role in beige adipogenesis via Th2 cytokines, influencing the generation and maturation of adipocyte precursors for beige adipocyte differentiation, and supporting thermogenic activation to improve energy metabolism.
THE REGULATION OF ADIPOKINES BY TH2 CYTOKINES IN ADIPOSE TISSUE
Emerging evidence supports a role of Th2 cytokines as a surrogate factor to stimulate the proliferation of M2 macrophages associated with low-grade inflammation in AT and differentiation of adipose precursors in metabolic homeostasis. A number of studies have established other effects of Th2 cytokines, including IL-33, IL-4, and IL-13, on adipokine secretion by adipocytes.
Accordingly, adipokines are considered to be regulators of whole-body homeostasis [71]. In addition to leptin, TNF-α, IL-6, resistin, FGF21, and adiponectin, which have been extensively reviewed, and a number of other key adipokines are involved in the regulation of inflammation [72737475]. We showed that growth differentiation factor 15 (GDF15) is a useful predictive biomarker of cardiovascular risk in newly diagnosed T2D patients [767778]. In addition, we identified the significance of neuregulin 4 (Nrg4) as a biomarker that is positively correlated with serum glucose level and insulin resistance in type 2 diabetes mellitus patients [77]. Kang et al. [78] reported significant increases in proinflammatory cytokine levels, such as monocyte chemoattractant protein-1 and TNF-α, in the VAT of patients with modest obesity and early metabolic dysfunction. Serum levels of adiponectin and leptin were significantly associated with insulin resistance and obesity. However, there were no obvious changes in macrophage phenotype or macrophage infiltration in patients with modest obesity or early metabolic dysfunction [78]. We showed that the expression of CD163/CD68, a marker of macrophage infiltration, was significantly related to mitochondrial biogenesis-associated genes, such as proliferator-activated receptor-γ coactivator 1α (PGC-1α), PGC-1β, and OXPHOS. Previous studies have also suggested that alternative activation of FAO and mitochondrial biogenesis in macrophages by PGC-1β may be related to obesity [7980]. Consequently, these findings suggest that the regulation of M2 macrophages in obesity may be closely linked to mitochondrial biogenesis, and that changes in the production of inflammatory biomolecules precede increased immune cell infiltration and induction of a macrophage phenotype switch in modest obesity in humans.
To explore the effects of Th2 cytokines on adipokines in AT homeostasis, we examined Th2 cytokine-mediated release of adipokines [81]. In vivo administration of α-galactosylceramide or IL-33 increased IL-4 and IL-13 production, thereby increasing GDF15 levels in AT and plasma in a STAT6-dependent manner using a STAT6 knockout mouse model. In addition, we found that administration of recombinant IL-13 to wild-type mice fed a high-fat diet improved glucose intolerance. Using GDF15-knockout mice, we showed that this effect was dependent on GDF15 expression. We found that Th2 cytokines regulate GDF15, and our results suggested that GDF15 is required for Th2 cytokine-induced improvement of glucose intolerance in metabolic disease [81].
GDF15, also known as macrophage-inhibiting cytokine 1, belongs to the transforming growth factor-β superfamily and is highly expressed in the heart, liver, kidneys, and colon [39]. Recently, the metabolic effects of GDF15 were found to include regulation of appetite by the orphan receptor, GFRAL [8283]. Moreover, recent evidence supports an important role of GDF15 in peripheral organs, including the liver, WAT, and muscle; GDF15 regulates systemic glucose tolerance as a mitochondrial unfolded protein response-associated critical cell nonautonomous myomitokine by promoting oxidation and lipolysis [84858687]. Further research is needed to identify peripheral receptors of GDF15 to elucidate the previse role of GDF15 in metabolic homeostasis.
THE ROLE OF MITOCHONDRIAL OXIDATIVE FUNCTION IN REGULATION OF MACROPHAGE POLARIZATION AND SYSTEMIC ENERGY HOMEOSTASIS
Increased glycolysis has been observed in lipopolysaccharide-stimulated macrophages [83]. A series of recent studies demonstrated that commitment to Warburg metabolism equips macrophages to fulfill their effector functions, such as production of reactive oxygen species (ROS) or nitric oxide, phagocytosis, and secretion of inflammatory mediators in the context of bacterial infection [40]. Upon activation with proinflammatory stimuli, such as lipopolysaccharide or IFN-γ, macrophages undergo metabolic reprogramming and exhibit increased rates of glycolysis and decreased OXPHOS. By diverting adenosine triphosphate generation from OXPHOS to glycolysis, mitochondria become available for ROS production [88].
The type 2 cytokines, IL-4 and IL-13, promote maturation into alternatively activated macrophages, and this effect is dependent on STAT6 activation [5]. Upon IL-4 stimulation, macrophages increase fatty acid uptake, β-oxidation, and OXPHOS; these metabolic rearrangements are initiated by STAT6 and peroxisome PGC-1β [88]. Moreover, STAT6 and its associated transcription factors, including PPAR-γ, PPAR-δ, and PGC-1α, critically influence M2-like macrophages by increasing oxidative metabolism and mitochondrial biogenesis [6789]. Studies in mice with macrophage-specific deletion of PPAR-γ have shown that PPAR-γ is required for maturation of alternatively activated macrophages [89]. Inhibition of FAO in M2 macrophages with the CPT1 inhibitor, etomoxir, limits M2 activation in response to IL-4 [90]. Blocking oxidative metabolism not only impairs the development of an M2-like phenotype, but also drives the cells toward an M1-like state [45].
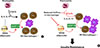
A recent study showed that blockade of lysosomal lipolysis during Heligmosomoides polygyrus infection results in defective clearance of the pathogen and inhibits commitment to OXPHOS by macrophages [90]. While changes in β-oxidation are the most striking metabolic adaptations in response to IL-4, metabolomic studies have revealed that glycolysis and glutaminolysis also contribute to TCA activity [91]. Recent research has focused on whether the oxidative function of macrophages is reduced under these conditions, and if so, whether such impairment induces insulin resistance. Using a mouse model, we found that defective oxidative function in macrophages due to myeloid-specific loss-of-function of the CR6-interacting factor 1 (Crif1) gene, an essential mitoribosomal factor required for biogenesis of OXPHOS subunits, results in systemic insulin resistance associated with adipose inflammation [92]. Moreover, macrophages from these mice are deficient in the release of GDF15, which is required for oxidative metabolism in M2-like macrophages stimulated with IL-4 and the PPAR-γ agonist, rosiglitazone. In addition, GDF15-deficient macrophages underwent polarization into an M1-like phenotype, and reintroduction of GDF15-null macrophages into high-fat diet-fed mice in which macrophages were depleted with clodronate resulted in glucose intolerance. We found that the mitochondrial oxidative function of macrophages has an important role in GDF15 secretion, and dysfunction of mitochondria in macrophages causes systemic insulin resistance and AT inflammation due to macrophage-regulated autocrine and paracrine signaling that promotes anti-inflammatory responses in WAT (Fig. 2).
CONCLUSIONS AND PERSPECTIVES
We have highlighted the role of type 2 immunity in metabolic disease. Recent studies have suggested that glucose intolerance and obesity induce the dysregulation of type 2 immunity. In addition, many studies have shown that type 2 immunity has a major influence on AT metabolism, including local effects, such as regulating AT browning, AT remodeling, and limiting excessive type 1 inflammatory responses, as well as systemic effects mediated via adipocytokines. Th2 cytokines, including IL-33, IL-4, and IL-13, have been identified as surrogate factors that stimulate the proliferation of M2 macrophages associated with low-grade inflammation in AT and differentiation of adipose precursors in metabolic homeostasis. Recently, we identified the roles of the Th2 cytokine (IL-4 and IL-13)-induced adipokine, GDF15, in AT in regulating systemic glucose metabolism via STAT6 activation. Moreover, we showed that mitochondrial OXPHOS is required to maintain these macrophage-regulating autocrine and paracrine signaling pathways via Th2 cytokine-induced secretion of GDF15. However, less is known regarding the mechanism of action of GDF15 in metabolic organs, such as muscle, liver, and AT. Interactive roles of Th2 cytokines and GDF15 must be further clarified by identification of the molecular action and signaling pathways of GDF15.
 XML Download
XML Download