

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
In β-cells of pancreas and liver, glucose transport machinery consists of glucose transporter type 2 (GLUT2) and glucokinase (Gck), which is also called a glucose sensor [1]. But in the 1980s, the mechanism of the regulation of gene expression of hepatic glucose transport system in the insulin resistant, diabetic states was largely unknown. Thus, we started to clone upstream regions (promoters) of GLUT2, Gck, and glucose-6-phosphatase (G6Pase) and searched for potential trans-acting factors regulating the expression of these genes. In this review, I will introduce my research path which has mainly focused on the characterization of the transcriptional network of genes involved in the glucose homeostasis.
Go to : 
STARTING FROM RAT GLUT2 PROMOTER
In the 1990s, cloning of genes with upstream regulatory elements was the mainstream biochemistry field. Because it was the previous era of the Genome Project we had to manually sequence a lot of short gene fragments using Sanger's dideoxy method to clone the whole rat GLUT2 gene, which was laborious and time consuming. Dr. Gil-Soo Han, my first research associate, started the project, but left my Lab in 1993 before the work was completed. So, we had to determine sequence of exon 1 with 5′-flanking region. Repeated screenings of lambda phage clones finally revealed the exon 1 and upstream sequences, and furthermore, using rapid amplification of 5′ complementary DNA ends (5′-RACE) and primer extension, we were able to identify complete structure of rat GLUT2 gene, which has four split exon 1 (1a to 1d). This discovery let us to obtain the rat GLUT2 promoter region, which conveys many potential transcription regulatory elements [2]. Next, we tried to find the mechanism of regulation of GLUT2 gene expression in liver. GLUT2 is a major glucose transporter in liver and pancreatic β-cells, and we initially focused to liver because nuclear extract from this organ is easier to prepare to study DNA-protein interaction, including DNase I footprinting and electrophoretic mobility shift assay (EMSA). The DNase I footprinting assay was a difficult and laborious experiment and took a long time, but finally we demonstrated CCAAT/enhancer binding protein (C/EBP) elements in the GLUT2 promoter. Furthermore, it was also identified that the regulation by C/EBPs were gradually attenuated in the primary cultured hepatocytes, providing an evidence why primary hepatocytes on culture dishes lose their ability to express GLUT2 [3]. These earlier studies became foundation stone of my research.
Go to :
EXPANSION TO HUMAN GLUT2 PROMOTER
GLUT2 gene is mainly expressed in liver and pancreatic β-cells; however, the transcription factors and cis-elements regulating the tissue-specific expression of the GLUT2 were largely unknown. Transfection studies using pancreatic β-cells (HIT-T15, MIN6), hepatoma cell (HepG2), and fibroblast cells (NIH3T3, HeLa) revealed the differences in the promoter activities and site C (87 to 132) responsible for tissue-specific expression of the human GLUT2 gene [4]. We identified that both hepatocyte nuclear factor 1 (HNF1) and HNF3 function as transcriptional activators in GLUT2 gene expression. The GLUT2 mRNA level was well correlated with the status of HNF1 and HNF3 mRNA levels regardless of the origins or types of cells. Furthermore, we showed that the promoter activity of the mutG construct containing the 103A→G mutation, which was found in a Korean non-insulin-dependent diabetes mellitus (NIDDM) patients, was in part compensated by the binding of nuclear factor Y (NF-Y) instead of HNF1 or HNF3 [5].
Go to :
NUCLEAR RECEPTOR AND GLUCOSE SENSOR REGULATION IN LIVER AND β-CELLS
In the late 1990s, peroxisome proliferator-activated receptor γ (PPARγ) was identified as a target for thiazolidinedione (TZD), the first insulin sensitizing agent [6]. Anti-diabetic action of TZD was thought to be mediated by PPARγ in adipose tissues until we identified that TZD restores the GLUT2 expression in pancreatic β-cells of type 2 diabetes mellitus (T2DM) model mice. While searching for transcription factors regulating GLUT2 gene, we noticed the presence of PPARγ response element (PPRE) in 5′ untranslated region of the GLUT2 gene. Although PPARγ in β-cells is not abundant, TZD treatment directly up-regulated PPARγ-dependent GLUT2 expression and restored glucose sensing ability of β-cell in diabetic condition [7]. Subsequently, we identified PPRE in β-cell specific glucokinase (βGck) promoter and showed its functional relevance in β-cells [8].
After that, we studied liver glucokinase (L-Gck) promoter, because we wanted to stay focused in the hepatic glucose sensor in connection with glucose homeostasis. In liver, we were able to identify and characterize the role of PPARγ in the regulation of L-Gck [910]. Next, we demonstrated the interrelationship among PPARγ, liver X-receptor α (LXRα), sterol regulatory element binding protein-1c (SREBP-1c), and small heterodimer partner (SHP) in the regulation of hepatic glucokinase expression [11].
Go to :
SREBP-1c AND GLUCOSE METABOLISM
The regulation of hepatic glucose metabolism is important in glucose homeostasis and L-Gck plays a central role in this process. L-Gck expression is known to be regulated by insulin and SREBP-1c was suggested to be a transcription factor that mediates the action of insulin on Gck transcription in liver. However, the mechanism how SREBP-1c induce hepatic GcK expression in response to insulin is not well characterized. Using DNA-protein interactions techniques, we identified sterol regulatory elements (SREs) in rat L-Gck promoter and present evidences that insulin increases the binding of SREBP-1c on L-Gck promoter resulting in the increase of the L-Gck transcription [12].
In 2004, we performed an experiment whether SREBP-1c could be a potential trans-acting factor regulating GLUT2 in liver. At that time, GLUT2 gene expression was not known to be regulated by insulin. And thus, SREBP-1c could not be a transcriptional coactivator of GLUT2 gene expression. However, we observed that both GLUT2 and SREBP-1c gene expression were increased in hyperglycemic states. Moreover, we demonstrated that SREBP-1c binding region (SRE) is present in the mouse GLUT2 promoter. With these data, we concluded that glucose-stimulated activation of GLUT2 gene expression might be induced by SREBP-1c in primary cultured hepatocytes. Adenoviral expression of the dominant negative form of SREBP-1c suppressed glucose-stimulated GLUT2 mRNA level in primary cultured hepatocytes [13].
We also showed regulatory mechanism of GLUT4 gene expression in adipocytes. With increased GLUT4 expression, mRNA levels of SREBP-1c in adipose tissue were proportionally increased by refeeding. Insulin treatment increased mRNA levels of GLUT4 in adipose tissue. Because SREBP-1c gene expression is regulated by insulin, it is thought that GLUT4 might be regulated by SREBP-1c. On this basis, we examined presence of a potential site binding SBREP-1c in the GLUT4 promoter and found highly conserved SRE. Through this study, we published a paper entitled, “Insulin-mediated activation of human GLUT4 promoter is directly induced by SREBP-1c in adipocytes” [14].
Subsequent to this study, we hypothesized that transcription factor E3 (TFE3) could be able to up-regulate Gck because TFE3 was shown to up-regulate genes of insulin signaling pathway [15]. Using adenoviral transduction of TFE3, we demonstrated the presence of TFE3 binding site (E-box) in the Gck and TFE3 up-regulates Gck with amelioration of hyperglycemia in the db/db and ob/ob mice [16]. Fig. 1 shows a summary of cis-elements and trans-acting factors acting on the glucose sensor genes in liver explored by my team.
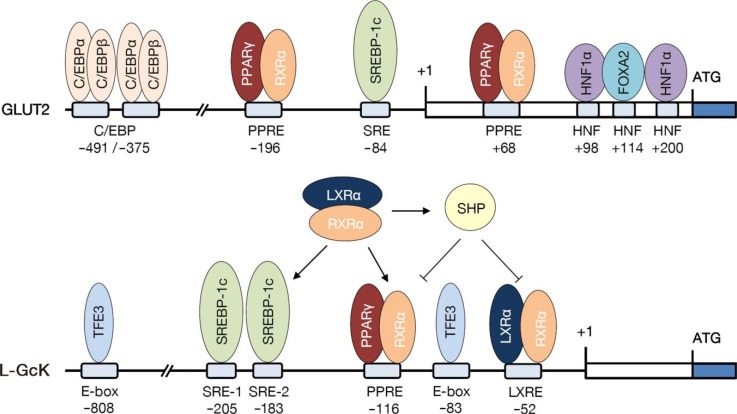 | Fig. 1Schematic summary of the regulation of liver type glucose transporter (glucose transporter type 2 [GLUT2]) and glucokinase (Gck) promoter. During the long journey to understand glucose homeostasis, main interest of my group was how glucose sensors, GLUT2 and glucokinase, are regulated by transcription factors. Tissue-specific study, and disease-specific alteration as well, revealed that many transcriptional factors are interconnected in the regulation of glucose sensor in liver. C/EBP, CCAAT/enhancer binding protein; PPARγ, peroxisome proliferator-activated receptor γ; RXRα, retinoid X receptor α; PPRE, PPARγ response element; SREBP-1c, sterol regulatory element binding protein-1c; SRE, sterol regulatory element; HNF1α, hepatocyte nuclear factor 1 α; HNF, hepatocyte nuclear factor; FOXA2, forkhead box A2; LXRα, liver X-receptor α; SHP, small heterodimer partner; L-GcK, liver glucokinase; TFE3, transcription factor E3; LXRE, liver X-receptor response element.
|
Go to :
ROLE OF PPARα AND PROLACTIN REGULATORY ELEMENT-BINDING ON THE GLUCONEOGENESIS
When glucose level is low, blood glucose level is maintained by hepatic gluconeogenesis. Because increase in a hepatic glucose production occurs in the post-absorptive state of T2DM, it is required to unveil the mechanisms of how hyperglycemia develops. To explore a pathogenesis, we are interested in a role of PPARα and prolactin regulatory element-binding (PREB). First, we have performed experiments based on the previous observation that PPARα-null mice shows severe hypoglycemia following 24-hour fasting, characterized by a 50% drop in blood glucose level, suggesting a potential role of PPARα in glucose metabolism [17]. Using PPARα knockout (KO) mice as an animal model, we were able to demonstrate presence of PPRE and indicated that PPARα might be responsible for glucose production through the regulation of hepatic G6pase expression [18]. Most recently, we observed that PREB protein is down-regulated in the liver of db/db, ob/ob, and high-fat diet-induced obese mice. This observation led us to explore a possibility that PREB could be a transacting factor down-regulating gluconeogenic. Indeed, we found a cis-element, prolactin core-binding element in the promoters of gluconeogenic genes, which is responsible for down-regulation of these genes, that is, decreased PREB in the liver of insulin resistant animal cannot repress the expression of gluconeogenic genes (G6Pase and phosphoenolpyruvate carboxykinase [Pck]), resulting in the hyperglycemia, a typical manifestation of the diabetic model mice or high fat induced obesity mice [19].
Go to :
ROLE OF PROTEIN INTERACTIONS AND GLUCOSE HOMEOSTASIS
Glucose homeostasis is of prime importance in maintaining whole body metabolism. In the insulin resistant states, a balance in the intermediary metabolism may be disturbed. During prolonged starvation, hepatic gluconeogenesis is a major pathway that maintains normal blood glucose levels. In the postprandial state, temporarily increased glucose needs to be disposed by liver to prevent the cells from glucotoxicity. In this process, Gck is known to play the most important role [20].
Although we have been stayed focus on the transcriptional aspects of metabolic genes in liver, we have also searched for the possibility that post-translational modification of metabolic enzymes/protein could also be a novel regulatory mechanism. Using current proteome technology, we were able to demonstrate that glucokinase regulatory protein (GKRP) in liver is acetylated by acetyltransferase p300. Acetylated GKRP decreases ubiquitination of the protein itself with an increase in its affinity to Gck, resulting in increased nuclear retention of Gck and decreased glycolytic flux [21].
To understand this, we have done couple of studies to elucidate mechanisms of how hepatic gluconeogenesis occurs. From the beginning, we showed that insulin-mediated repression of G6pase and Pck gene expression is restored by resveratrol, which might be associated with sirtuin 1 (Sirt1) mediated forkhead box O1 (FOXO1) deacetylation [22]. And then, we found a role of thioredoxin-interacting protein (TXNIP) in the hyperglycemia. The increased TXNIP in the hyperglycemic mice model, impairs glucose and insulin tolerance in mice by up-regulating G6pase through interaction with SHP. Binding of TXNIP to SHP resulted in the degradation of SHP through ubiquitination, allowing FOXO1 to up-regulate gluconeogenic gene expression [23].
Go to :
ROLE OF CREB3L4 ON THE ADIPOGENESIS
Another field of our research was transcriptional connections in adipogenesis. We were interested in obesity problem, which became a worldwide epidemic. To this end, we wanted to explore signaling networks governed by transcription factors involved in the adipogenesis, ultimately to understand the pathophysiology of obesity and T2DM. Using cyclic AMP-responsive element-binding protein 3 (CREB3)-like 4 (CREB3L4) KO mice, we demonstrated that CREB3L4 acts as a negative regulator of adipogenesis [24]. In this paper we demonstrated that CREB3L4 binds C/EBPβ, promoting its ubiquitination, resulting in the inhibition of adipogenesis and obesity in mice (Fig. 2A). Thus, if a measure to trigger CREB3L4 expression is taken, it could be one of novel way to treat obesity or T2DM. In connection with this work, we published a paper entitled, “The effects of low fat diet (LFD) or aging on the metabolic profiles of Creb3l4 KO mice,” which showed significant weight gain and adiposity with impaired glucose tolerance and decreased insulin sensitivity in Creb3l4 KO mice [25]. Furthermore, we demonstrated that CREB3L4 is required for proliferation of prostate cancer cells and CREB3L4 is a crucial activator of androgen receptor (AR) function. CREB3L4 directly interacts with, and facilitates, AR recruitment to the androgen responsive elements (AREs) of AR target genes. In addition, indirectly, through inositol requiring enzyme 1α signaling, a distinct AR-endoplasmic reticulum stress-CREB3L4 regulatory axis also plays a role in prostate cancer proliferation (Fig. 2B) [26]. Table 1 shows a summary of major works done by my team for the last 25 years.
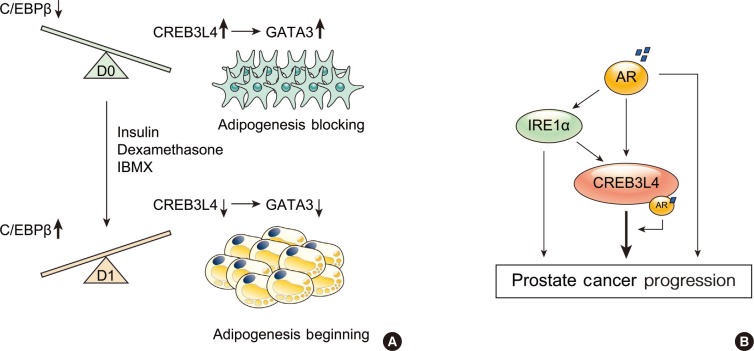 | Fig. 2Role of cyclic AMP-responsive element-binding protein 3 (CREB3)-like 4 (CREB3L4) on the adipogenesis. (A) In preadipocytes, CREB3L4 acts as a negative regulator of adipogenesis by both regulating the stability of CCAAT/enhancer binding protein β (C/EBPβ) protein and increasing GATA binding protein 3 (GATA3) expression. (B) Proposed mechanism of action of CREB3L4 in prostate cancer progression. Adapted from Kim et al. [2426]. IBMX, 3-isobutyl-1-methylxanthine; AR, androgen receptor; IRE1α, inositol requiring enzyme 1α.
|
Table 1
Summary of major research papers of my team
| Year | Journal | Research subject | Contributor(s) |
|---|---|---|---|
| 1995 | Arch Biochem Biophys | L-GLUT2 promoter cloning | Ahn et al. [2] |
| 1998 | Biochem J | L-GLUT2 promoter, C/EBP | Kim et al. [3] |
| 2000 | J Biol Chem | L-GLUT2 liver specific expression, HNF1&3 | Cha et al. [4] |
| 2000 | Diabetes | L-GLUT2 promoter, PPRE | Kim et al. [7] |
| 2002 | Diabetes | Β-Glucokinase, PPARγ | Kim et al. [8] |
| 2004 | Diabetes | L-GcK promoter, PPARγ | Kim et al. [10] |
| 2004 | J Biol Chem | L-GcK promoter, SREBP-1c | Kim et al. [12] |
| 2005 | Diabetes | L-GLUT2 promoter, SREBP-1c | Im et al. [13] |
| 2006 | Biochem J | GLUT4 promoter, SREBP-1c adipocytes | Im et al. [14] |
| 2009 | J Biol Chem | L-GcK promoter, LXR, SREBP-1c, PPARγ | Kim et al. [11] |
| 2011 | J Biol Chem | G6Pase promoter, PPARα | Im et al. [18] |
| 2013 | Diabetologia | L-GcK gene, TFE3 | Kim et al. [16] |
| 2013 | Diabetologia | Liver gluconeogenic gene, Txnip | Jo et al. [23] |
| 2014 | Cell Death Dis | Adipogenesis and CREB3L4 | Kim et al. [24] |
| 2015 | Sci Rep | L-GcK and GKRP acetylation | Park et al. [21] |
| 2017 | Sci Rep | Prostate cancer and CREB3L4 | Kim et al. [26] |
| 2018 | BBA Mol Basis Dis | Liver PREB and glucose homeostasis | Park et al. [19] |
L-GLUT2, liver-glucose transporter type 2; C/EBP, CCAAT/enhancer binding protein; HNF, hepatocyte nuclear factor; PPRE, PPARγ response element; PPAR, peroxisome proliferator-activated receptor; L-GcK, liver glucokinase; SREBP-1c, sterol regulatory element binding protein-1c; LXR, liver X-receptor; G6Pase, glucose-6-phosphatase; TFE3, transcription factor E3; CREB3L4, cyclic AMP-responsive element-binding protein 3 (CREB3)-like 4; GKRP, glucokinase regulatory protein; PREB, prolactin regulatory element-binding.
![]()
Go to :
SUMMARY
My team has been exploring the roles of transcription factors regulating the genes involved in glucose metabolism for the last 30 years. The main field of study can be summarized as (1) identification of promoters of genes of glucose homeostasis, (2) exploration of interaction between promoters and transcription factors leading to tissue-specific gene expression, (3) modification and interactions between transcription factors in metabolic diseases such as diabetes or obesity, and (4) role of a transcription factor in the adipocyte differentiation and development of obesity. While studying transcriptional regulation, my team members were interconnected as if we were transcription factors coordinating with each other. As an analogy, I might act as a basal transcription factor and my team-mates, wherever they are, played co-activator role. Without their devotion in science, the works of my group could not be made possible.
Go to :
 XML Download
XML Download